{"title":"WEDAP: A Python Package for Streamlined Plotting of Molecular Simulation Data","authors":"Darian T. Yang, and , Lillian T. Chong*, ","doi":"10.1021/acs.jcim.4c0086710.1021/acs.jcim.4c00867","DOIUrl":null,"url":null,"abstract":"<p >Given the growing interest in path sampling methods for extending the time scales of molecular dynamics (MD) simulations, there has been great interest in software tools that streamline the generation of plots for monitoring the progress of large-scale simulations. Here, we present the WEDAP Python package for simplifying the analysis of data generated from either conventional MD simulations or the weighted ensemble (WE) path sampling method, as implemented in the widely used WESTPA software package. WEDAP facilitates (i) the parsing of WE simulation data stored in highly compressed, hierarchical HDF5 files and (ii) incorporates trajectory weights from WE simulations into all generated plots. Our Python package consists of multiple user-friendly interfaces: a command-line interface, a graphical user interface, and a Python application programming interface. We demonstrate the plotting features of WEDAP through a series of examples using data from WE and conventional MD simulations that focus on the HIV-1 capsid protein’s C-terminal domain dimer as a showcase system. The source code for WEDAP is freely available on GitHub at https://github.com/chonglab-pitt/wedap.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 15","pages":"5749–5755 5749–5755"},"PeriodicalIF":5.3000,"publicationDate":"2024-07-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jcim.4c00867","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00867","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
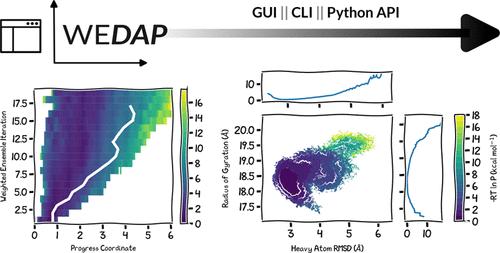
Given the growing interest in path sampling methods for extending the time scales of molecular dynamics (MD) simulations, there has been great interest in software tools that streamline the generation of plots for monitoring the progress of large-scale simulations. Here, we present the WEDAP Python package for simplifying the analysis of data generated from either conventional MD simulations or the weighted ensemble (WE) path sampling method, as implemented in the widely used WESTPA software package. WEDAP facilitates (i) the parsing of WE simulation data stored in highly compressed, hierarchical HDF5 files and (ii) incorporates trajectory weights from WE simulations into all generated plots. Our Python package consists of multiple user-friendly interfaces: a command-line interface, a graphical user interface, and a Python application programming interface. We demonstrate the plotting features of WEDAP through a series of examples using data from WE and conventional MD simulations that focus on the HIV-1 capsid protein’s C-terminal domain dimer as a showcase system. The source code for WEDAP is freely available on GitHub at https://github.com/chonglab-pitt/wedap.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


