Density functional theory guide for copolymerization mechanism between allyl radical with radicalophile: photo-driven radical mediated [3 + 2] cyclization
Ou Liu, Piaoyi Chen, Qinglin Xiao, Chengfeng Yue, Yugang Huang, Guodong Ye
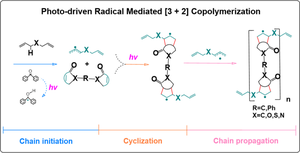
{"title":"Density functional theory guide for copolymerization mechanism between allyl radical with radicalophile: photo-driven radical mediated [3 + 2] cyclization","authors":"Ou Liu, Piaoyi Chen, Qinglin Xiao, Chengfeng Yue, Yugang Huang, Guodong Ye","doi":"10.1007/s00894-024-06104-x","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>The challenge of activating inert allyl monomers for polymerization has persisted, prompting our proposal of the photo-driven radical mediated [3 + 2] cyclization reaction (PRMC). This innovative approach significantly expedites the homopolymerization of multi-allyl monomers, enabling the synthesis of embolic microspheres for hepatocellular carcinoma interventions. PRMC involves allyl monomers to form allylic radicals and then radicals participating in a cycloaddition reaction with unsaturated olefins as radicalophiles to form cyclopentane-based radical products. While extensively studied in the theoretical and experimental homopolymerization, PRMC’s application in copolymerization remains unexplored. To address this knowledge gap, we explored the elementary reaction, selecting allyl methyl ether radicals (AMER) and α,β-unsaturated ketones as radicalophiles for copolymerization investigations by density functional theory (DFT) analysis. We quantified energy differences between ground and excited states of reactants, elucidated frontier molecular orbitals, and assessed thermodynamic data for copolymerization feasibility. We also evaluated the electronic properties of reactants, predicting the reactivity of radicalophiles and the interactions of intermolecular reactions. Additionally, we applied transition state theory and interaction/deformation models and conducted a local orbital analysis to comprehensively study excess electron distribution and gyration radius of cyclic radical product. Our findings offer vital insights into PRMC’s potential in copolymerization. This research provides a robust theoretical foundation for practical application, enhancing the polymerization field.</p><h3>Methods</h3><p>Based on density functional theory (DFT), the calculations were performed at the M06-2X/6–311 + + G(d,p) level in/by Gaussian 16 package. Subsequently, our analytical results apply time-dependent density-functional theory (TD-DFT) and solvent modeling (SMD). Single-point energy calculations determine the driving force behind the radicals’ reaction with radicalophiles. Furthermore, we assessed the electrostatic potential (ESP) of the reactants. The results of the calculations were visualized by the Multiwfn 3.6 and VMD 1.9 programs.</p><h3>Graphical abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":null,"pages":null},"PeriodicalIF":2.1000,"publicationDate":"2024-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06104-x","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
The challenge of activating inert allyl monomers for polymerization has persisted, prompting our proposal of the photo-driven radical mediated [3 + 2] cyclization reaction (PRMC). This innovative approach significantly expedites the homopolymerization of multi-allyl monomers, enabling the synthesis of embolic microspheres for hepatocellular carcinoma interventions. PRMC involves allyl monomers to form allylic radicals and then radicals participating in a cycloaddition reaction with unsaturated olefins as radicalophiles to form cyclopentane-based radical products. While extensively studied in the theoretical and experimental homopolymerization, PRMC’s application in copolymerization remains unexplored. To address this knowledge gap, we explored the elementary reaction, selecting allyl methyl ether radicals (AMER) and α,β-unsaturated ketones as radicalophiles for copolymerization investigations by density functional theory (DFT) analysis. We quantified energy differences between ground and excited states of reactants, elucidated frontier molecular orbitals, and assessed thermodynamic data for copolymerization feasibility. We also evaluated the electronic properties of reactants, predicting the reactivity of radicalophiles and the interactions of intermolecular reactions. Additionally, we applied transition state theory and interaction/deformation models and conducted a local orbital analysis to comprehensively study excess electron distribution and gyration radius of cyclic radical product. Our findings offer vital insights into PRMC’s potential in copolymerization. This research provides a robust theoretical foundation for practical application, enhancing the polymerization field.
Methods
Based on density functional theory (DFT), the calculations were performed at the M06-2X/6–311 + + G(d,p) level in/by Gaussian 16 package. Subsequently, our analytical results apply time-dependent density-functional theory (TD-DFT) and solvent modeling (SMD). Single-point energy calculations determine the driving force behind the radicals’ reaction with radicalophiles. Furthermore, we assessed the electrostatic potential (ESP) of the reactants. The results of the calculations were visualized by the Multiwfn 3.6 and VMD 1.9 programs.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


