Miguel Cosenza-Contreras, Adrianna Seredynska, Daniel Vogele, Niko Pinter, Eva Brombacher, Ruth Fiestas Cueto, Thien-Ly Julia Dinh, Patrick Bernhard, Manuel Rogg, Junwei Liu, Patrick Willems, Simon Stael, Pitter F. Huesgen, E. Wolfgang Kuehn, Clemens Kreutz, Christoph Schell, Oliver Schilling
{"title":"TermineR: Extracting information on endogenous proteolytic processing from shotgun proteomics data","authors":"Miguel Cosenza-Contreras, Adrianna Seredynska, Daniel Vogele, Niko Pinter, Eva Brombacher, Ruth Fiestas Cueto, Thien-Ly Julia Dinh, Patrick Bernhard, Manuel Rogg, Junwei Liu, Patrick Willems, Simon Stael, Pitter F. Huesgen, E. Wolfgang Kuehn, Clemens Kreutz, Christoph Schell, Oliver Schilling","doi":"10.1002/pmic.202300491","DOIUrl":null,"url":null,"abstract":"<p>State-of-the-art mass spectrometers combined with modern bioinformatics algorithms for peptide-to-spectrum matching (PSM) with robust statistical scoring allow for more variable features (i.e., post-translational modifications) being reliably identified from (tandem-) mass spectrometry data, often without the need for biochemical enrichment. Semi-specific proteome searches, that enforce a theoretical enzymatic digestion to solely the N- or C-terminal end, allow to identify of native protein termini or those arising from endogenous proteolytic activity (also referred to as “neo-N-termini” analysis or “N-terminomics”). Nevertheless, deriving biological meaning from these search outputs can be challenging in terms of data mining and analysis. Thus, we introduce <i>TermineR</i>, a data analysis approach for the (1) annotation of peptides according to their enzymatic cleavage specificity and known protein processing features, (2) differential abundance and enrichment analysis of N-terminal sequence patterns, and (3) visualization of neo-N-termini location. We illustrate the use of <i>TermineR</i> by applying it to tandem mass tag (TMT)-based proteomics data of a mouse model of polycystic kidney disease, and assess the semi-specific searches for biological interpretation of cleavage events and the variable contribution of proteolytic products to general protein abundance. The <i>TermineR</i> approach and example data are available as an R package at https://github.com/MiguelCos/TermineR.</p>","PeriodicalId":224,"journal":{"name":"Proteomics","volume":"24 19","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-08-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/pmic.202300491","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomics","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/pmic.202300491","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
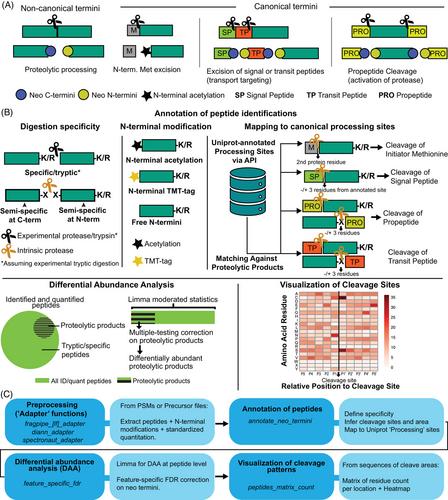
State-of-the-art mass spectrometers combined with modern bioinformatics algorithms for peptide-to-spectrum matching (PSM) with robust statistical scoring allow for more variable features (i.e., post-translational modifications) being reliably identified from (tandem-) mass spectrometry data, often without the need for biochemical enrichment. Semi-specific proteome searches, that enforce a theoretical enzymatic digestion to solely the N- or C-terminal end, allow to identify of native protein termini or those arising from endogenous proteolytic activity (also referred to as “neo-N-termini” analysis or “N-terminomics”). Nevertheless, deriving biological meaning from these search outputs can be challenging in terms of data mining and analysis. Thus, we introduce TermineR, a data analysis approach for the (1) annotation of peptides according to their enzymatic cleavage specificity and known protein processing features, (2) differential abundance and enrichment analysis of N-terminal sequence patterns, and (3) visualization of neo-N-termini location. We illustrate the use of TermineR by applying it to tandem mass tag (TMT)-based proteomics data of a mouse model of polycystic kidney disease, and assess the semi-specific searches for biological interpretation of cleavage events and the variable contribution of proteolytic products to general protein abundance. The TermineR approach and example data are available as an R package at https://github.com/MiguelCos/TermineR.
最先进的质谱仪与现代生物信息学算法相结合,利用强大的统计评分功能进行肽谱匹配(PSM),可以从(串联)质谱数据中可靠地识别出更多可变特征(即翻译后修饰),通常无需进行生化富集。半特异性蛋白质组搜索只对 N 端或 C 端进行理论上的酶解,可识别原生蛋白质末端或由内源性蛋白水解活动产生的蛋白质末端(也称为 "新 N 端 "分析或 "N 端组学")。然而,要从这些搜索结果中得出生物学意义,在数据挖掘和分析方面具有挑战性。因此,我们引入了 TermineR,这是一种数据分析方法,用于(1)根据酶裂解特异性和已知蛋白质加工特征对肽段进行注释,(2)对 N 端序列模式进行差异丰度和富集分析,以及(3)对新 N 端位置进行可视化。我们将 TermineR 应用于基于串联质量标签 (TMT) 的多囊肾小鼠模型蛋白质组学数据,以此说明 TermineR 的用途,并评估半特异性搜索对裂解事件的生物学解释以及蛋白水解产物对一般蛋白质丰度的不同贡献。TermineR方法和示例数据作为R软件包可在https://github.com/MiguelCos/TermineR。
期刊介绍:
PROTEOMICS is the premier international source for information on all aspects of applications and technologies, including software, in proteomics and other "omics". The journal includes but is not limited to proteomics, genomics, transcriptomics, metabolomics and lipidomics, and systems biology approaches. Papers describing novel applications of proteomics and integration of multi-omics data and approaches are especially welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


