Factor V haemostatic diathesis impairing thrombin activation, membrane binding and circulating antigen level due to a novel compound heterozygous mutation, Leu1821Ser and Gly2192Cys
Kimberley Talbot, Jina Song, John R. Perrier, Shannon Jackson, Ross T. A. MacGillivray, Edward L. G. Pryzdial
{"title":"Factor V haemostatic diathesis impairing thrombin activation, membrane binding and circulating antigen level due to a novel compound heterozygous mutation, Leu1821Ser and Gly2192Cys","authors":"Kimberley Talbot, Jina Song, John R. Perrier, Shannon Jackson, Ross T. A. MacGillivray, Edward L. G. Pryzdial","doi":"10.1111/hae.15087","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Introduction</h3>\n \n <p>Congenital factor V (FV) deficiency is a rare clotting disorder affecting ∼1 in 1,000,000, with bleeding severity that ranges broadly for poorly understood reasons.</p>\n </section>\n \n <section>\n \n <h3> Aim</h3>\n \n <p>To help understand the molecular basis of the observed phenotype in FV deficient patients, the genetics and biochemistry causing a patient's FV deficiency were evaluated.</p>\n </section>\n \n <section>\n \n <h3> Methods and Results</h3>\n \n <p>A 71-year-old female, who had serious life-long bleeding upon provocation and profound menorrhagia that lead to hysterectomy, was found to have 3% of normal plasma FV antigen with normal electrophoretic mobility. Platelet FV was similarly low, although the banding pattern was less fragmented than normal. Plasma clotting activity was <1% of normal. Familial inheritance and DNA sequence analysis from peripheral blood leukocytes were consistent with novel compound heterozygosity with missense mutations in exon XVII, Leu1821 to Ser (L1821S) and exon XXV, Gly2192 to Cys (G2192C). The respective single-mutation variants were expressed and purified. Explaining why the antigen level and activity were inequivalent, thrombin activation of recombinant (<i>r</i>) FV/L1821S was impaired, and rFV/G2192C was unable to bind to a procoagulant phospholipid membrane.</p>\n </section>\n \n <section>\n \n <h3> Conclusion</h3>\n \n <p>These findings are consistent with the observed phenotype, highlighting the importance of understanding FV biochemical function to rationalize clinical bleeding severity when the circulating antigen level is discordant.</p>\n </section>\n </div>","PeriodicalId":12819,"journal":{"name":"Haemophilia","volume":"30 5","pages":"1170-1176"},"PeriodicalIF":3.0000,"publicationDate":"2024-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/hae.15087","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Haemophilia","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/hae.15087","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction
Congenital factor V (FV) deficiency is a rare clotting disorder affecting ∼1 in 1,000,000, with bleeding severity that ranges broadly for poorly understood reasons.
Aim
To help understand the molecular basis of the observed phenotype in FV deficient patients, the genetics and biochemistry causing a patient's FV deficiency were evaluated.
Methods and Results
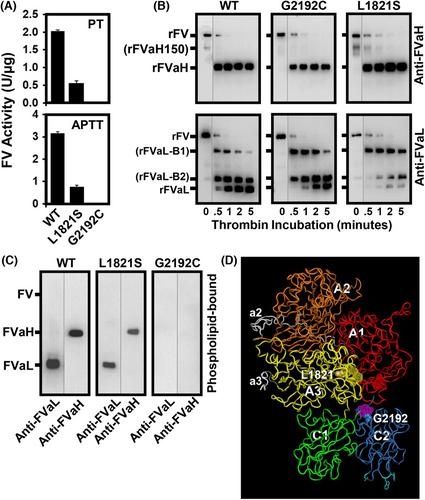
A 71-year-old female, who had serious life-long bleeding upon provocation and profound menorrhagia that lead to hysterectomy, was found to have 3% of normal plasma FV antigen with normal electrophoretic mobility. Platelet FV was similarly low, although the banding pattern was less fragmented than normal. Plasma clotting activity was <1% of normal. Familial inheritance and DNA sequence analysis from peripheral blood leukocytes were consistent with novel compound heterozygosity with missense mutations in exon XVII, Leu1821 to Ser (L1821S) and exon XXV, Gly2192 to Cys (G2192C). The respective single-mutation variants were expressed and purified. Explaining why the antigen level and activity were inequivalent, thrombin activation of recombinant (r) FV/L1821S was impaired, and rFV/G2192C was unable to bind to a procoagulant phospholipid membrane.
Conclusion
These findings are consistent with the observed phenotype, highlighting the importance of understanding FV biochemical function to rationalize clinical bleeding severity when the circulating antigen level is discordant.
期刊介绍:
Haemophilia is an international journal dedicated to the exchange of information regarding the comprehensive care of haemophilia. The Journal contains review articles, original scientific papers and case reports related to haemophilia care, with frequent supplements. Subjects covered include:
clotting factor deficiencies, both inherited and acquired: haemophilia A, B, von Willebrand''s disease, deficiencies of factor V, VII, X and XI
replacement therapy for clotting factor deficiencies
component therapy in the developing world
transfusion transmitted disease
haemophilia care and paediatrics, orthopaedics, gynaecology and obstetrics
nursing
laboratory diagnosis
carrier detection
psycho-social concerns
economic issues
audit
inherited platelet disorders.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


