Thionation-Induced Enhancement of Optical and Electronic Properties in NDI Molecule for Molecular Electronic Applications: A Computational Study Using DFT/TD-DFT and QTAIM Theory
{"title":"Thionation-Induced Enhancement of Optical and Electronic Properties in NDI Molecule for Molecular Electronic Applications: A Computational Study Using DFT/TD-DFT and QTAIM Theory","authors":"Hamid Hadi, Hamid Reza Shamlouei","doi":"10.1002/adts.202400151","DOIUrl":null,"url":null,"abstract":"<p>The study investigates the impact of thionation on N,N'-di(dodecyl)-4,5,8,9-naphthalene diimide (NDI) through computational methods such as density functional theory (DFT/TD-DFT), quantum theory of atoms in molecules (QTAIM), and Landauer theory (LT). Thionation, involving the replacement of diamide oxygens with sulfurs in NDI, significantly enhances quantum-electronic/thermoelectric properties. Computational analyzes of energy of frontier orbitals HOMO/LUMO, dipole moment, polarizability, first superpolarizability, UV spectrum, and cohesive energy show the superior performance of the thione structure (M2) compared to the pristine structure (M1). Thionation decreased the energy gap from 01.3 eV (in M1 structure) to 1.87 eV (in M2 structure). The absorption wavelength in the pristine structure (M1) is calculated to be 507 nm, which increased to 1067 nm after thionation (M2). Cohesive energy values for each of M1 and M2 structures are calculated as 12.76 and 12.89 Kcal mol<sup>−1</sup>, respectively, which indicates the improvement of stability after thionation. After connecting M1 and M2 to gold electrodes (Au-M1-Au and Au-M2-Au) and applying electric fields, the Au-M2-Au structure shows a lower energy gap, lower thermoelectric activity and higher conductivity at field intensities with higher than 140 × 10<sup>−4</sup> (a.u.), indicating its use as a field-effect molecular device (such as molecular wire or molecular switch).</p>","PeriodicalId":7219,"journal":{"name":"Advanced Theory and Simulations","volume":"7 11","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2024-08-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advanced Theory and Simulations","FirstCategoryId":"5","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/adts.202400151","RegionNum":4,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
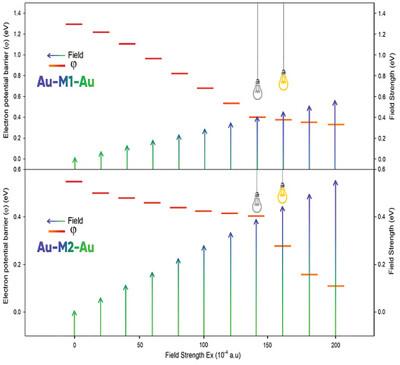
The study investigates the impact of thionation on N,N'-di(dodecyl)-4,5,8,9-naphthalene diimide (NDI) through computational methods such as density functional theory (DFT/TD-DFT), quantum theory of atoms in molecules (QTAIM), and Landauer theory (LT). Thionation, involving the replacement of diamide oxygens with sulfurs in NDI, significantly enhances quantum-electronic/thermoelectric properties. Computational analyzes of energy of frontier orbitals HOMO/LUMO, dipole moment, polarizability, first superpolarizability, UV spectrum, and cohesive energy show the superior performance of the thione structure (M2) compared to the pristine structure (M1). Thionation decreased the energy gap from 01.3 eV (in M1 structure) to 1.87 eV (in M2 structure). The absorption wavelength in the pristine structure (M1) is calculated to be 507 nm, which increased to 1067 nm after thionation (M2). Cohesive energy values for each of M1 and M2 structures are calculated as 12.76 and 12.89 Kcal mol−1, respectively, which indicates the improvement of stability after thionation. After connecting M1 and M2 to gold electrodes (Au-M1-Au and Au-M2-Au) and applying electric fields, the Au-M2-Au structure shows a lower energy gap, lower thermoelectric activity and higher conductivity at field intensities with higher than 140 × 10−4 (a.u.), indicating its use as a field-effect molecular device (such as molecular wire or molecular switch).
期刊介绍:
Advanced Theory and Simulations is an interdisciplinary, international, English-language journal that publishes high-quality scientific results focusing on the development and application of theoretical methods, modeling and simulation approaches in all natural science and medicine areas, including:
materials, chemistry, condensed matter physics
engineering, energy
life science, biology, medicine
atmospheric/environmental science, climate science
planetary science, astronomy, cosmology
method development, numerical methods, statistics

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


