Francesco E. Emiliani, Donald C. Green, Edward G. Hughes, Wahab A. Khan, George J. Zanazzi, Chun-Chieh Lin
{"title":"A pituitary mass in a 46-year-old woman","authors":"Francesco E. Emiliani, Donald C. Green, Edward G. Hughes, Wahab A. Khan, George J. Zanazzi, Chun-Chieh Lin","doi":"10.1111/bpa.13295","DOIUrl":null,"url":null,"abstract":"<p>A 46-year-old woman presented to the clinic with a 6-week history of left retro-orbital headaches. In recent weeks, she had developed increased thirst, a salty taste, and diplopia with blurry vision in the left eye. On examination, left cranial nerve six palsy was noted. Magnetic resonance imaging (MRI) revealed a 2.7 cm heterogeneously enhancing pituitary mass (Figure 1). Laboratory findings showed slightly elevated prolactin (83.8 ng/mL; reference: 4.8–23.3 ng/mL). There were no other findings associated with hypo/hyperpituitarism. During transsphenoidal resection, the tumor appeared to be adherent and firm with indistinct boundary with the pituitary gland. A portion of the tumor infiltrating the cavernous sinus was left behind to prevent damage to the cavernous segment of the carotid artery.</p><p>Intraoperative cytological smears showed pleomorphic tumor cells with prominent nucleoli and minimal cytoplasm (Figure 2A and Box 1). Histological examination of formalin-fixed paraffin-embedded (FFPE) tissue showed a poorly differentiated neoplasm with high mitotic counts (up to five per high-power field), forming a sheet-like architecture. Tumor cells exhibited prominent nucleoli and vesicular chromatin with cytoplasmic vacuolation. Occasional intracytoplasmic globular eosinophilic inclusions were noted (Figure 2B). Immunohistochemical study showed that the tumor was negative for CK (AE1/AE3), GFAP, synaptophysin, LCA, Melan A, HMB-45, Oct3/4, PLAP, and brachyury. Notably, the tumor showed loss of nuclear SMARCB1/INI1 staining (Figure 2C). Ki67 index was elevated (70%).</p><p>Chromosomal microarray analysis (CMA) demonstrated copy neutral loss of heterozygosity (cnLOH) in nearly the entire chromosome 22 that includes the <i>SMARCB1</i> locus at 22q11.2 (Figure 2D). Next-generation sequencing (NGS) detected a frameshift deletion in the 3′ end of <i>SMARCB1</i> (p. P383Rfs c.1148del; VAF = 74.2%) (Figure 2E). No other mutation was identified. Brain tumor methylation profiling from National Cancer Institute classified this tumor as an AT/RT subclass MYC (calibrated score of 0.95).</p><p>Adult sellar atypical teratoid/rhabdoid tumor, CNS WHO grade 4.</p><p>Adult sellar AT/RT is a rare, aggressive CNS embryonal tumor, with less than 50 cases reported to date. By definition, it occurs in the sellar/suprasellar region of adults, displays variable polyphenotypic differentiation and demonstrates characteristic <i>SMARCB1</i> mutations. Notably, there is a strong female predominance (95%) and middle age distribution (mean = 44 ± 3 years) [<span>1-3</span> and current case]. This contrasts with pediatric AT/RT, which shows a slight male predominance and has not been reported in the sellar region.</p><p>On MRI, adult sellar AT/RT is frequently described as a heterogeneously enhancing pituitary mass with invasion of adjacent structures. Due to its rarity and non-specific histological findings, the differential diagnosis is usually broad [<span>1-3</span>]. A pathological diagnosis should be considered when encountering a poorly differentiated neoplasm with rhabdoid features and frequent mitoses in the sellar region of female adults. In such cases, before extensive immunohistochemical studies, SMARCB1/INI1 immunostaining and molecular study should be prioritized to avoid specimen exhaustion. Adult sellar AT/RT shows characteristic nuclear SMARCB1/INI1 protein loss. Subsequent evaluation generally aims to identify polyphenotypic differentiation and to rule out other possibilities, such as pituitary adenoma/pituitary neuroendocrine tumor (synaptophysin), metastasis (cytokeratin, SMA, vimentin), rhabdoid meningioma (EMA, PR), germ cell tumor (PLAP and Oct3/4), lymphoma (LCA) and glioblastoma (GFAP). At the clivus/skull base, SMARCB1/INI1 protein loss could be detected in poorly differentiated chordomas with epithelioid and focal rhabdoid morphology. However, it typically retains brachyury expression and occurs in different demographic (median age of 11 years; female-to-male ratio of 2). Moreover, unlike AT/RT, the mechanism of SMARCB1/INI1 loss in chordomas involves homozygous deletion of <i>SMARCB1</i> gene.</p><p>Molecular and cytogenetic analysis are critical for accurate diagnosis. In AT/RT, genetic alterations often include a mutation in one allele of <i>SMARCB1</i>, followed by a second allele loss due to monosomy 22, deletion of 22q11.2, or an acquired cnLOH [<span>1</span>]. Our case exhibits a cytosine deletion in the exon 9 of <i>SMARCB1</i> (c.1148del) with concurrent cnLOH of chromosome 22. Interestingly, instead of introducing a premature stop codon and truncated protein, this deletion leads to a frameshift mutation in the last three codons, resulting in the addition of 99 extra amino acids before termination. Mechanistically, the elongated protein is predicted to be inactive, but how this results in loss of SMARCB1/INI1 expression, as shown by negative immunostaining, requires further investigation.</p><p>Finally, methylation profiling of pediatric ATRT has established three distinct subtypes based on the overexpression of melanosomal genes (TYR), sonic hedgehog pathway (SHH), and MYC and HOX cluster. Consistent with recent findings that seven adult sellar AT/RTs are classified as ATRT-MYC subtype [<span>2</span>], our case was classified as an ATRT-MYC subtypes by the NIH classifier.</p><p>FEE and CCL wrote the draft. DCG, EGH, and WAK analyzed the data. CCL and GJZ did histological and immunohistochemical analysis. All authors review and approve the final version of the manuscript.</p><p>The authors declare no conflicts of interest.</p><p>All data related to the case are deidentified.</p>","PeriodicalId":9290,"journal":{"name":"Brain Pathology","volume":"34 5","pages":""},"PeriodicalIF":6.2000,"publicationDate":"2024-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/bpa.13295","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain Pathology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/bpa.13295","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
A 46-year-old woman presented to the clinic with a 6-week history of left retro-orbital headaches. In recent weeks, she had developed increased thirst, a salty taste, and diplopia with blurry vision in the left eye. On examination, left cranial nerve six palsy was noted. Magnetic resonance imaging (MRI) revealed a 2.7 cm heterogeneously enhancing pituitary mass (Figure 1). Laboratory findings showed slightly elevated prolactin (83.8 ng/mL; reference: 4.8–23.3 ng/mL). There were no other findings associated with hypo/hyperpituitarism. During transsphenoidal resection, the tumor appeared to be adherent and firm with indistinct boundary with the pituitary gland. A portion of the tumor infiltrating the cavernous sinus was left behind to prevent damage to the cavernous segment of the carotid artery.
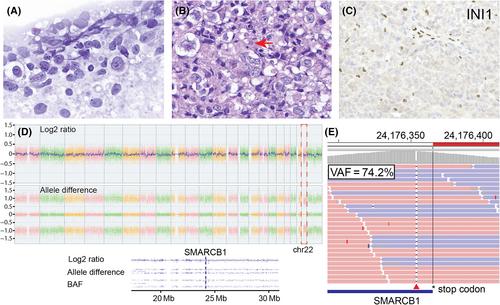
Intraoperative cytological smears showed pleomorphic tumor cells with prominent nucleoli and minimal cytoplasm (Figure 2A and Box 1). Histological examination of formalin-fixed paraffin-embedded (FFPE) tissue showed a poorly differentiated neoplasm with high mitotic counts (up to five per high-power field), forming a sheet-like architecture. Tumor cells exhibited prominent nucleoli and vesicular chromatin with cytoplasmic vacuolation. Occasional intracytoplasmic globular eosinophilic inclusions were noted (Figure 2B). Immunohistochemical study showed that the tumor was negative for CK (AE1/AE3), GFAP, synaptophysin, LCA, Melan A, HMB-45, Oct3/4, PLAP, and brachyury. Notably, the tumor showed loss of nuclear SMARCB1/INI1 staining (Figure 2C). Ki67 index was elevated (70%).
Chromosomal microarray analysis (CMA) demonstrated copy neutral loss of heterozygosity (cnLOH) in nearly the entire chromosome 22 that includes the SMARCB1 locus at 22q11.2 (Figure 2D). Next-generation sequencing (NGS) detected a frameshift deletion in the 3′ end of SMARCB1 (p. P383Rfs c.1148del; VAF = 74.2%) (Figure 2E). No other mutation was identified. Brain tumor methylation profiling from National Cancer Institute classified this tumor as an AT/RT subclass MYC (calibrated score of 0.95).
Adult sellar atypical teratoid/rhabdoid tumor, CNS WHO grade 4.
Adult sellar AT/RT is a rare, aggressive CNS embryonal tumor, with less than 50 cases reported to date. By definition, it occurs in the sellar/suprasellar region of adults, displays variable polyphenotypic differentiation and demonstrates characteristic SMARCB1 mutations. Notably, there is a strong female predominance (95%) and middle age distribution (mean = 44 ± 3 years) [1-3 and current case]. This contrasts with pediatric AT/RT, which shows a slight male predominance and has not been reported in the sellar region.
On MRI, adult sellar AT/RT is frequently described as a heterogeneously enhancing pituitary mass with invasion of adjacent structures. Due to its rarity and non-specific histological findings, the differential diagnosis is usually broad [1-3]. A pathological diagnosis should be considered when encountering a poorly differentiated neoplasm with rhabdoid features and frequent mitoses in the sellar region of female adults. In such cases, before extensive immunohistochemical studies, SMARCB1/INI1 immunostaining and molecular study should be prioritized to avoid specimen exhaustion. Adult sellar AT/RT shows characteristic nuclear SMARCB1/INI1 protein loss. Subsequent evaluation generally aims to identify polyphenotypic differentiation and to rule out other possibilities, such as pituitary adenoma/pituitary neuroendocrine tumor (synaptophysin), metastasis (cytokeratin, SMA, vimentin), rhabdoid meningioma (EMA, PR), germ cell tumor (PLAP and Oct3/4), lymphoma (LCA) and glioblastoma (GFAP). At the clivus/skull base, SMARCB1/INI1 protein loss could be detected in poorly differentiated chordomas with epithelioid and focal rhabdoid morphology. However, it typically retains brachyury expression and occurs in different demographic (median age of 11 years; female-to-male ratio of 2). Moreover, unlike AT/RT, the mechanism of SMARCB1/INI1 loss in chordomas involves homozygous deletion of SMARCB1 gene.
Molecular and cytogenetic analysis are critical for accurate diagnosis. In AT/RT, genetic alterations often include a mutation in one allele of SMARCB1, followed by a second allele loss due to monosomy 22, deletion of 22q11.2, or an acquired cnLOH [1]. Our case exhibits a cytosine deletion in the exon 9 of SMARCB1 (c.1148del) with concurrent cnLOH of chromosome 22. Interestingly, instead of introducing a premature stop codon and truncated protein, this deletion leads to a frameshift mutation in the last three codons, resulting in the addition of 99 extra amino acids before termination. Mechanistically, the elongated protein is predicted to be inactive, but how this results in loss of SMARCB1/INI1 expression, as shown by negative immunostaining, requires further investigation.
Finally, methylation profiling of pediatric ATRT has established three distinct subtypes based on the overexpression of melanosomal genes (TYR), sonic hedgehog pathway (SHH), and MYC and HOX cluster. Consistent with recent findings that seven adult sellar AT/RTs are classified as ATRT-MYC subtype [2], our case was classified as an ATRT-MYC subtypes by the NIH classifier.
FEE and CCL wrote the draft. DCG, EGH, and WAK analyzed the data. CCL and GJZ did histological and immunohistochemical analysis. All authors review and approve the final version of the manuscript.
期刊介绍:
Brain Pathology is the journal of choice for biomedical scientists investigating diseases of the nervous system. The official journal of the International Society of Neuropathology, Brain Pathology is a peer-reviewed quarterly publication that includes original research, review articles and symposia focuses on the pathogenesis of neurological disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


