Gökçe Geylan, Leonardo De Maria, Ola Engkvist, Florian David and Ulf Norinder
{"title":"A methodology to correctly assess the applicability domain of cell membrane permeability predictors for cyclic peptides†","authors":"Gökçe Geylan, Leonardo De Maria, Ola Engkvist, Florian David and Ulf Norinder","doi":"10.1039/D4DD00056K","DOIUrl":null,"url":null,"abstract":"<p >Being able to predict the cell permeability of cyclic peptides is essential for unlocking their potential as a drug modality for intracellular targets. With a wide range of studies of cell permeability but a limited number of data points, the reliability of the machine learning (ML) models to predict previously unexplored chemical spaces becomes a challenge. In this work, we systemically investigate the predictive capability of ML models from the perspective of their extrapolation to never-before-seen applicability domains, with a particular focus on the permeability task. Four predictive algorithms, namely Support-Vector Machine, Random Forest, LightGBM and XGBoost, jointly with a conformal prediction framework were employed to characterize and evaluate the applicability through uncertainty quantification. Efficiency and validity of the models' predictions with multiple calibration strategies were assessed with respect to several external datasets from different parts of the chemical space through a set of experiments. The experiments showed that the predictors generalizing well to the applicability domain defined by the training data, can fail to achieve similar model performance on other parts of the chemical spaces. Our study proposes an approach to overcome such limitations by the means of improving the efficiency of models without sacrificing the validity. The trade-off between the reliability and informativeness was balanced when the models were calibrated with a subset of the data from the new targeted domain. This study outlines an approach to enable the extrapolation of predictive power and restore the models' reliability <em>via</em> a recalibration strategy without the need for retraining the underlying model.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":" 9","pages":" 1761-1775"},"PeriodicalIF":6.2000,"publicationDate":"2024-07-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d4dd00056k?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d4dd00056k","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
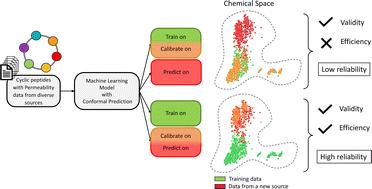
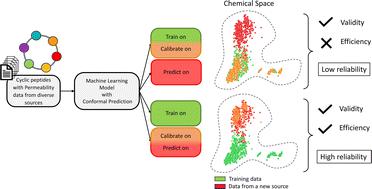
Being able to predict the cell permeability of cyclic peptides is essential for unlocking their potential as a drug modality for intracellular targets. With a wide range of studies of cell permeability but a limited number of data points, the reliability of the machine learning (ML) models to predict previously unexplored chemical spaces becomes a challenge. In this work, we systemically investigate the predictive capability of ML models from the perspective of their extrapolation to never-before-seen applicability domains, with a particular focus on the permeability task. Four predictive algorithms, namely Support-Vector Machine, Random Forest, LightGBM and XGBoost, jointly with a conformal prediction framework were employed to characterize and evaluate the applicability through uncertainty quantification. Efficiency and validity of the models' predictions with multiple calibration strategies were assessed with respect to several external datasets from different parts of the chemical space through a set of experiments. The experiments showed that the predictors generalizing well to the applicability domain defined by the training data, can fail to achieve similar model performance on other parts of the chemical spaces. Our study proposes an approach to overcome such limitations by the means of improving the efficiency of models without sacrificing the validity. The trade-off between the reliability and informativeness was balanced when the models were calibrated with a subset of the data from the new targeted domain. This study outlines an approach to enable the extrapolation of predictive power and restore the models' reliability via a recalibration strategy without the need for retraining the underlying model.
要挖掘环肽作为细胞内靶点药物模式的潜力,预测环肽的细胞渗透性至关重要。由于对细胞渗透性的研究范围广泛,但数据点数量有限,因此机器学习(ML)模型预测以前未探索过的化学空间的可靠性就成了一个挑战。在这项工作中,我们从外推法的角度系统地研究了 ML 模型对前所未见的应用领域的预测能力,并特别关注渗透性任务。我们采用了四种预测算法,即支持向量机、随机森林、LightGBM 和 XGBoost,并结合保形预测框架,通过不确定性量化来描述和评估其适用性。通过一系列实验,针对来自化学空间不同部分的多个外部数据集,评估了采用多种校准策略的模型预测的效率和有效性。实验结果表明,对训练数据所定义的适用性领域具有良好普适性的预测器,在化学空间的其他部分可能无法实现类似的模型性能。我们的研究提出了一种在不牺牲有效性的前提下提高模型效率的方法来克服这种局限性。当使用新目标领域的数据子集校准模型时,可靠性和信息量之间的权衡得到了平衡。本研究概述了一种通过重新校准策略实现预测能力外推并恢复模型可靠性的方法,而无需重新训练基础模型。

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


