{"title":"Benchmarking Methods for PROTAC Ternary Complex Structure Prediction.","authors":"Evianne Rovers, Matthieu Schapira","doi":"10.1021/acs.jcim.4c00426","DOIUrl":null,"url":null,"abstract":"<p><p>Proteolysis targeting chimeras (PROTACs) are bifunctional compounds that recruit an E3 ligase to a target protein to induce ubiquitination and degradation of the target. Rational optimization of PROTAC requires a structural model of the ternary complex. In the absence of an experimental structure, computational tools have emerged that attempt to predict PROTAC ternary complexes. Here, we systematically benchmark three commonly used tools: PRosettaC, MOE, and ICM. We find that these PROTAC-focused methods produce an array of ternary complex structures, including some that are observed experimentally, but also many that significantly deviate from the crystal structure. Molecular dynamics simulations show that PROTAC complexes may exist in a multiplicity of configurational states and question the use of experimentally observed structures as a reference for accurate predictions. The pioneering computational tools benchmarked here highlight the promises and challenges in the field and may be more valuable when guided by clear structural and biophysical data. The benchmarking data set that we provide may also be valuable for evaluating other and future computational tools for ternary complex modeling.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":null,"pages":null},"PeriodicalIF":5.6000,"publicationDate":"2024-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00426","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/1 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
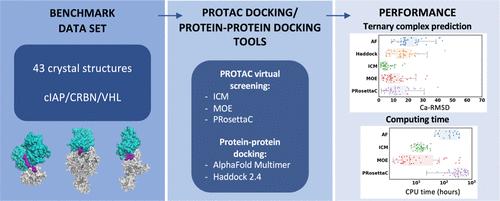
Abstract
Proteolysis targeting chimeras (PROTACs) are bifunctional compounds that recruit an E3 ligase to a target protein to induce ubiquitination and degradation of the target. Rational optimization of PROTAC requires a structural model of the ternary complex. In the absence of an experimental structure, computational tools have emerged that attempt to predict PROTAC ternary complexes. Here, we systematically benchmark three commonly used tools: PRosettaC, MOE, and ICM. We find that these PROTAC-focused methods produce an array of ternary complex structures, including some that are observed experimentally, but also many that significantly deviate from the crystal structure. Molecular dynamics simulations show that PROTAC complexes may exist in a multiplicity of configurational states and question the use of experimentally observed structures as a reference for accurate predictions. The pioneering computational tools benchmarked here highlight the promises and challenges in the field and may be more valuable when guided by clear structural and biophysical data. The benchmarking data set that we provide may also be valuable for evaluating other and future computational tools for ternary complex modeling.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


