M. Hamdi Cherif , L. Beldi , M. Houari , B. Bouadjemi , S. Bentata , S. Haid , M. Matougui , T. Lantri , B. Achour , S. Mesbah , A. Khatar , B. Bouhafs , N. Alnawmasi , W. Khalifa
{"title":"Investigating the multifaceted characteristics of Ba2FeWO6 double perovskite: Insights from density functional theory","authors":"M. Hamdi Cherif , L. Beldi , M. Houari , B. Bouadjemi , S. Bentata , S. Haid , M. Matougui , T. Lantri , B. Achour , S. Mesbah , A. Khatar , B. Bouhafs , N. Alnawmasi , W. Khalifa","doi":"10.1016/j.jmgm.2024.108834","DOIUrl":null,"url":null,"abstract":"<div><p>This study undertook a comprehensive examination of the double perovskite complex Ba<sub>2</sub>FeWO<sub>6</sub>, investigating its structural, electrical, magnetic, thermal and elastic characteristics. The study used density functional theory (DFT), specifically the full potential linearized augmented plane wave (FP-LAPW) method. It also used different approximations, including the generalized gradient approximation (GGA) and the modified Trans-Blaha (TB-mBJ) approach, to improve the accuracy of the band gap estimation more accurate. Additionlly, the GGA + U approach, incorporating the Hubbard correction term (U), was utilized. Our findings indicate that Ba<sub>2</sub>FeWO<sub>6</sub> exhibits indirect half-metallic band gaps in the (L-X) direction, with value of 0.91 eV and a net magnetic moment of 4 <em>μ</em><sub>B</sub>, predominatly influenced by the iron atom. The compound demonstrated exceptional characteristics suitable for thermoelectric applications, particularly at lower temperatures. Furthermore, the elasticity analysis revealed low brittleness, facilitates its manipulation in manufacturing procedures.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"132 ","pages":"Article 108834"},"PeriodicalIF":2.7000,"publicationDate":"2024-07-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001347","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
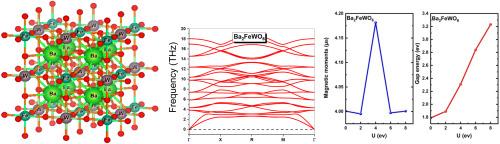
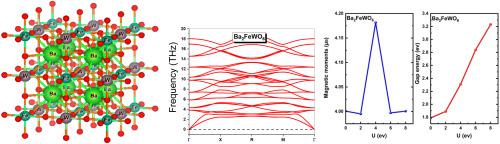
This study undertook a comprehensive examination of the double perovskite complex Ba2FeWO6, investigating its structural, electrical, magnetic, thermal and elastic characteristics. The study used density functional theory (DFT), specifically the full potential linearized augmented plane wave (FP-LAPW) method. It also used different approximations, including the generalized gradient approximation (GGA) and the modified Trans-Blaha (TB-mBJ) approach, to improve the accuracy of the band gap estimation more accurate. Additionlly, the GGA + U approach, incorporating the Hubbard correction term (U), was utilized. Our findings indicate that Ba2FeWO6 exhibits indirect half-metallic band gaps in the (L-X) direction, with value of 0.91 eV and a net magnetic moment of 4 μB, predominatly influenced by the iron atom. The compound demonstrated exceptional characteristics suitable for thermoelectric applications, particularly at lower temperatures. Furthermore, the elasticity analysis revealed low brittleness, facilitates its manipulation in manufacturing procedures.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


