{"title":"COSMOPharm: Drug-Polymer Compatibility of Pharmaceutical Amorphous Solid Dispersions from COSMO-SAC.","authors":"Ivan Antolović, Jadran Vrabec, Martin Klajmon","doi":"10.1021/acs.molpharmaceut.4c00342","DOIUrl":null,"url":null,"abstract":"<p><p>The quantum mechanics-aided COSMO-SAC activity coefficient model is applied and systematically examined for predicting the thermodynamic compatibility of drugs and polymers. The drug-polymer compatibility is a key aspect in the rational selection of optimal polymeric carriers for pharmaceutical amorphous solid dispersions (ASD) that enhance drug bioavailability. The drug-polymer compatibility is evaluated in terms of both solubility and miscibility, calculated using standard thermodynamic equilibrium relations based on the activity coefficients predicted by COSMO-SAC. As inherent to COSMO-SAC, our approach relies only on quantum-mechanically derived σ-profiles of the considered molecular species and involves no parameter fitting to experimental data. All σ-profiles used were determined in this work, with those of the polymers being derived from their shorter oligomers by replicating the properties of their central monomer unit(s). Quantitatively, COSMO-SAC achieved an overall average absolute deviation of 13% in weight fraction drug solubility predictions compared to experimental data. Qualitatively, COSMO-SAC correctly categorized different polymer types in terms of their compatibility with drugs and provided meaningful estimations of the amorphous-amorphous phase separation. Furthermore, we analyzed the sensitivity of the COSMO-SAC results for ASD to different model configurations and σ-profiles of polymers. In general, while the free volume and dispersion terms exerted a limited effect on predictions, the structures of oligomers used to produce σ-profiles of polymers appeared to be more important, especially in the case of strongly interacting polymers. Explanations for these observations are provided. COSMO-SAC proved to be an efficient method for compatibility prediction and polymer screening in ASD, particularly in terms of its performance-cost ratio, as it relies only on first-principles calculations for the considered molecular species. The open-source nature of both COSMO-SAC and the Python-based tool COSMOPharm, developed in this work for predicting the API-polymer thermodynamic compatibility, invites interested readers to explore and utilize this method for further research or assistance in the design of pharmaceutical formulations.</p>","PeriodicalId":52,"journal":{"name":"Molecular Pharmaceutics","volume":" ","pages":"4395-4415"},"PeriodicalIF":4.5000,"publicationDate":"2024-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11372840/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Pharmaceutics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1021/acs.molpharmaceut.4c00342","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/30 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
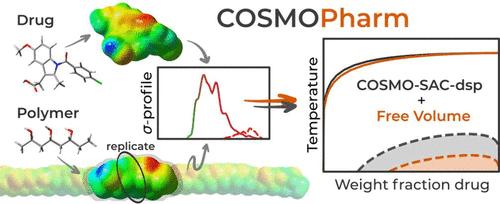
Abstract
The quantum mechanics-aided COSMO-SAC activity coefficient model is applied and systematically examined for predicting the thermodynamic compatibility of drugs and polymers. The drug-polymer compatibility is a key aspect in the rational selection of optimal polymeric carriers for pharmaceutical amorphous solid dispersions (ASD) that enhance drug bioavailability. The drug-polymer compatibility is evaluated in terms of both solubility and miscibility, calculated using standard thermodynamic equilibrium relations based on the activity coefficients predicted by COSMO-SAC. As inherent to COSMO-SAC, our approach relies only on quantum-mechanically derived σ-profiles of the considered molecular species and involves no parameter fitting to experimental data. All σ-profiles used were determined in this work, with those of the polymers being derived from their shorter oligomers by replicating the properties of their central monomer unit(s). Quantitatively, COSMO-SAC achieved an overall average absolute deviation of 13% in weight fraction drug solubility predictions compared to experimental data. Qualitatively, COSMO-SAC correctly categorized different polymer types in terms of their compatibility with drugs and provided meaningful estimations of the amorphous-amorphous phase separation. Furthermore, we analyzed the sensitivity of the COSMO-SAC results for ASD to different model configurations and σ-profiles of polymers. In general, while the free volume and dispersion terms exerted a limited effect on predictions, the structures of oligomers used to produce σ-profiles of polymers appeared to be more important, especially in the case of strongly interacting polymers. Explanations for these observations are provided. COSMO-SAC proved to be an efficient method for compatibility prediction and polymer screening in ASD, particularly in terms of its performance-cost ratio, as it relies only on first-principles calculations for the considered molecular species. The open-source nature of both COSMO-SAC and the Python-based tool COSMOPharm, developed in this work for predicting the API-polymer thermodynamic compatibility, invites interested readers to explore and utilize this method for further research or assistance in the design of pharmaceutical formulations.
期刊介绍:
Molecular Pharmaceutics publishes the results of original research that contributes significantly to the molecular mechanistic understanding of drug delivery and drug delivery systems. The journal encourages contributions describing research at the interface of drug discovery and drug development.
Scientific areas within the scope of the journal include physical and pharmaceutical chemistry, biochemistry and biophysics, molecular and cellular biology, and polymer and materials science as they relate to drug and drug delivery system efficacy. Mechanistic Drug Delivery and Drug Targeting research on modulating activity and efficacy of a drug or drug product is within the scope of Molecular Pharmaceutics. Theoretical and experimental peer-reviewed research articles, communications, reviews, and perspectives are welcomed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


