{"title":"Fully Flexible Molecular Alignment Enables Accurate Ligand Structure Modeling.","authors":"Zhihao Wang, Fan Zhou, Zechen Wang, Qiuyue Hu, Yong-Qiang Li, Sheng Wang, Yanjie Wei, Liangzhen Zheng, Weifeng Li, Xiangda Peng","doi":"10.1021/acs.jcim.4c00669","DOIUrl":null,"url":null,"abstract":"<p><p>Accurate protein-ligand binding poses are the prerequisites of structure-based binding affinity prediction and provide the structural basis for in-depth lead optimization in small molecule drug design. However, it is challenging to provide reasonable predictions of binding poses for different molecules due to the complexity and diversity of the chemical space of small molecules. Similarity-based molecular alignment techniques can effectively narrow the search range, as structurally similar molecules are likely to have similar binding modes, with higher similarity usually correlated to higher success rates. However, molecular similarity is not consistently high because molecules often require changes to achieve specific purposes, leading to reduced alignment precision. To address this issue, we propose a new alignment method─Z-align. This method uses topological structural information as a criterion for evaluating similarity, reducing the reliance on molecular fingerprint similarity. Our method has achieved success rates significantly higher than those of other methods at moderate levels of similarity. Additionally, our approach can comprehensively and flexibly optimize bond lengths and angles of molecules, maintaining a high accuracy even when dealing with larger molecules. Consequently, our proposed solution helps in achieving more accurate binding poses in protein-ligand docking problems, facilitating the development of small molecule drugs. Z-align is freely available as a web server at https://cloud.zelixir.com/zalign/home.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":null,"pages":null},"PeriodicalIF":5.6000,"publicationDate":"2024-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00669","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/29 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
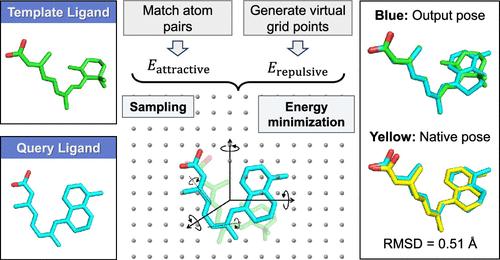
Abstract
Accurate protein-ligand binding poses are the prerequisites of structure-based binding affinity prediction and provide the structural basis for in-depth lead optimization in small molecule drug design. However, it is challenging to provide reasonable predictions of binding poses for different molecules due to the complexity and diversity of the chemical space of small molecules. Similarity-based molecular alignment techniques can effectively narrow the search range, as structurally similar molecules are likely to have similar binding modes, with higher similarity usually correlated to higher success rates. However, molecular similarity is not consistently high because molecules often require changes to achieve specific purposes, leading to reduced alignment precision. To address this issue, we propose a new alignment method─Z-align. This method uses topological structural information as a criterion for evaluating similarity, reducing the reliance on molecular fingerprint similarity. Our method has achieved success rates significantly higher than those of other methods at moderate levels of similarity. Additionally, our approach can comprehensively and flexibly optimize bond lengths and angles of molecules, maintaining a high accuracy even when dealing with larger molecules. Consequently, our proposed solution helps in achieving more accurate binding poses in protein-ligand docking problems, facilitating the development of small molecule drugs. Z-align is freely available as a web server at https://cloud.zelixir.com/zalign/home.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


