Asifa Kalwar, Quratulain Rid, F. N. U. Sadia, Sidhant Ochani
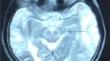
{"title":"Wilson’s Disease Masquerading as Acute Encephalitis: A Case Report","authors":"Asifa Kalwar, Quratulain Rid, F. N. U. Sadia, Sidhant Ochani","doi":"10.1007/s42399-024-01711-4","DOIUrl":null,"url":null,"abstract":"<p>Wilson’s disease (WD) is a rare autosomal recessive disorder characterized by copper accumulation in the liver, brain, and cornea, resulting from mutations in the ATPase copper transporting beta (ATP7B) gene. This case report presents a unique manifestation of WD, as a 14-year-old female from rural Pakistan presented with acute encephalitis and the “Face of the Giant Panda Sign.” Encephalitis is an uncommon initial presentation of WD, and this case highlights the diagnostic challenges, especially in resource-limited settings. The patient exhibited sudden-onset fever, generalized tonic–clonic seizures, and altered mental status. Initially misattributed to possession by the parents, the patient underwent empirical treatment for encephalitis in the hospital without improvement. Further evaluation revealed abnormal liver function, positive hepatitis B surface antigen (HepBsAg), and characteristic brain magnetic resonance imaging (MRI) findings indicative of WD. Confirmation was made through low ceruloplasmin, Kayser-Fleischer rings, and modified Leipzig criteria. Treatment with penicillamine and vitamin B6 resulted in clinical improvement. This case emphasizes the importance of considering WD in cases of encephalitis, even in the absence of typical hepatic symptoms. The delay in diagnosis and initial misinterpretation underscore the need for increased awareness and education, particularly in regions with limited healthcare resources. Early recognition and intervention can prevent the progression of WD, improving patient outcomes. The report also underscores the significance of familial screening and long-term follow-up to manage the chronic nature of the disease. Further studies and reporting of atypical presentations contribute to the understanding and management of this rare and clinically heterogeneous disorder.</p>","PeriodicalId":21944,"journal":{"name":"SN Comprehensive Clinical Medicine","volume":"415 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-07-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"SN Comprehensive Clinical Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s42399-024-01711-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Wilson’s disease (WD) is a rare autosomal recessive disorder characterized by copper accumulation in the liver, brain, and cornea, resulting from mutations in the ATPase copper transporting beta (ATP7B) gene. This case report presents a unique manifestation of WD, as a 14-year-old female from rural Pakistan presented with acute encephalitis and the “Face of the Giant Panda Sign.” Encephalitis is an uncommon initial presentation of WD, and this case highlights the diagnostic challenges, especially in resource-limited settings. The patient exhibited sudden-onset fever, generalized tonic–clonic seizures, and altered mental status. Initially misattributed to possession by the parents, the patient underwent empirical treatment for encephalitis in the hospital without improvement. Further evaluation revealed abnormal liver function, positive hepatitis B surface antigen (HepBsAg), and characteristic brain magnetic resonance imaging (MRI) findings indicative of WD. Confirmation was made through low ceruloplasmin, Kayser-Fleischer rings, and modified Leipzig criteria. Treatment with penicillamine and vitamin B6 resulted in clinical improvement. This case emphasizes the importance of considering WD in cases of encephalitis, even in the absence of typical hepatic symptoms. The delay in diagnosis and initial misinterpretation underscore the need for increased awareness and education, particularly in regions with limited healthcare resources. Early recognition and intervention can prevent the progression of WD, improving patient outcomes. The report also underscores the significance of familial screening and long-term follow-up to manage the chronic nature of the disease. Further studies and reporting of atypical presentations contribute to the understanding and management of this rare and clinically heterogeneous disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


