{"title":"Density functional theory study on frustrated Lewis pairs catalyzed C-H activation of heteroarenes: Mechanism variation tuning by electronic effect","authors":"Youxiang Shao, Kang Xiao, Huize Wang, Yalan Liu","doi":"10.1002/poc.4652","DOIUrl":null,"url":null,"abstract":"<p>Unreactive C-H bond activation is a new horizon for frustrated Lewis pairs (FLPs) chemistry. Although concerted mechanism (Science 2015, 349, 513) and stepwise carbene mechanism (Org. Lett. 2018, 20, 1102) have been proposed for the FLPs catalyzed C-H bond activation of 1-methylpyrrole, the influence of electronic properties of FLPs on the reaction mechanism is far away from well-understood. In this study, an assortment of P-B type FLPs with different electronic characteristic was employed to study the catalyzed C-H bond activation of 1-methylpyrrole by using density functional theory calculations. Detailed calculations demonstrated that the reactivity variation and the reaction mechanism binary of FLPs catalyzed C-H activation can be varied by tuning electronic effect of Lewis base center. On the one hand, the concerted C-H activation reactivity is mainly controlled by the electron donation of the lone pair of Lewis base center; thus, the FLPs with electron-donating substituents <b>(FLP1</b>, <b>FLP2</b>, and <b>FLP3)</b> catalyzed the C-H bond activation through concerted mechanism. On the other hand, the reactivity of stepwise carbene mechanism is mostly attributed to the vacant orbital of Lewis acid center; as a result, the <b>FLP5</b> bearing -P(C<sub>6</sub>F<sub>5</sub>)<sub>2</sub> preferred to catalyzed the bond activation through concerted mechanism. In contrast, a metathesis mechanism through strained four-membered ring transition state is less feasible. These results should provide deeper insight and broader perspective to understand the structure and function of FLPs for rational design of FLPs catalyzed C-H bond activation.</p>","PeriodicalId":16829,"journal":{"name":"Journal of Physical Organic Chemistry","volume":"37 10","pages":""},"PeriodicalIF":1.9000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physical Organic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/poc.4652","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
引用次数: 0
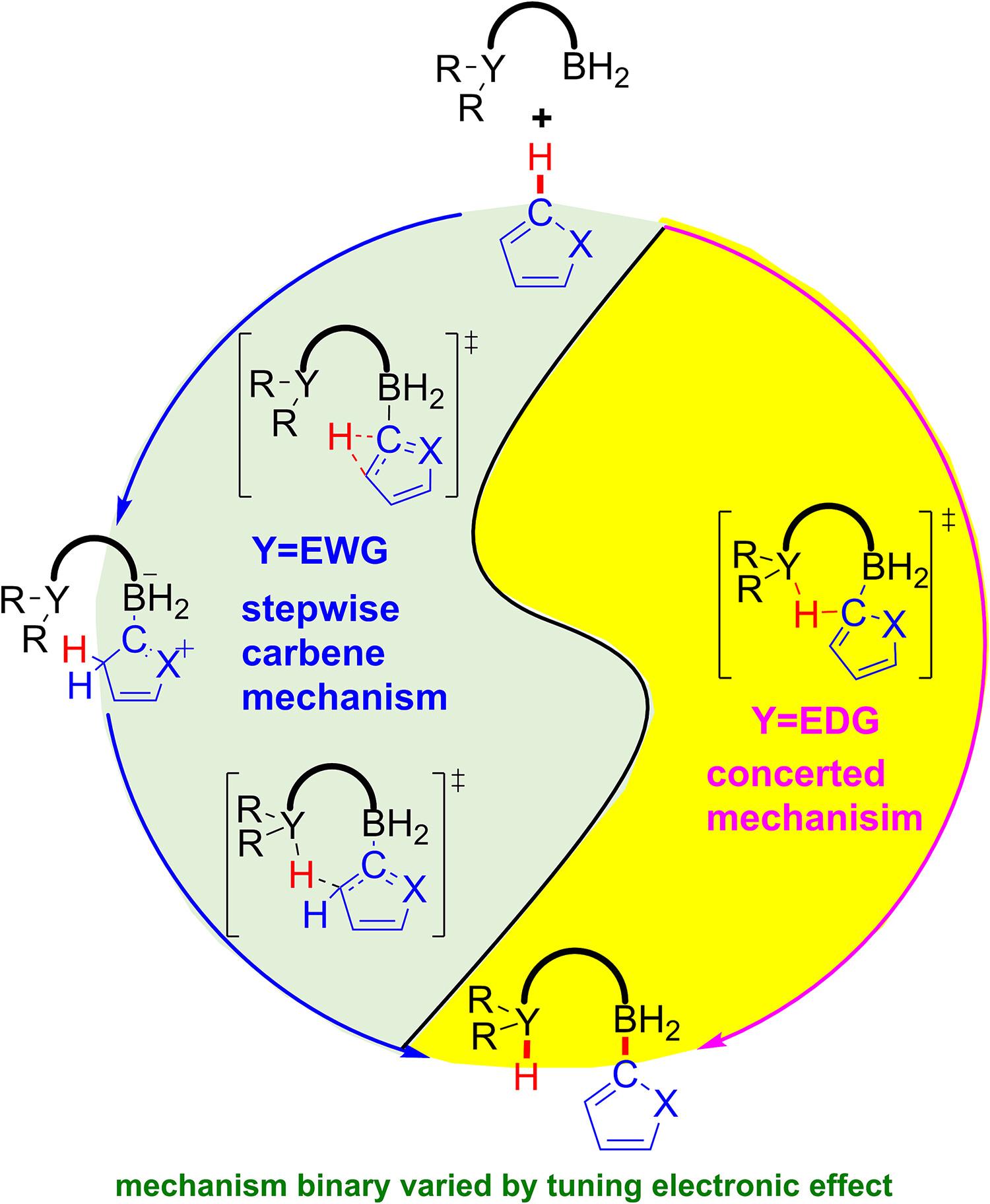
Abstract
Unreactive C-H bond activation is a new horizon for frustrated Lewis pairs (FLPs) chemistry. Although concerted mechanism (Science 2015, 349, 513) and stepwise carbene mechanism (Org. Lett. 2018, 20, 1102) have been proposed for the FLPs catalyzed C-H bond activation of 1-methylpyrrole, the influence of electronic properties of FLPs on the reaction mechanism is far away from well-understood. In this study, an assortment of P-B type FLPs with different electronic characteristic was employed to study the catalyzed C-H bond activation of 1-methylpyrrole by using density functional theory calculations. Detailed calculations demonstrated that the reactivity variation and the reaction mechanism binary of FLPs catalyzed C-H activation can be varied by tuning electronic effect of Lewis base center. On the one hand, the concerted C-H activation reactivity is mainly controlled by the electron donation of the lone pair of Lewis base center; thus, the FLPs with electron-donating substituents (FLP1, FLP2, and FLP3) catalyzed the C-H bond activation through concerted mechanism. On the other hand, the reactivity of stepwise carbene mechanism is mostly attributed to the vacant orbital of Lewis acid center; as a result, the FLP5 bearing -P(C6F5)2 preferred to catalyzed the bond activation through concerted mechanism. In contrast, a metathesis mechanism through strained four-membered ring transition state is less feasible. These results should provide deeper insight and broader perspective to understand the structure and function of FLPs for rational design of FLPs catalyzed C-H bond activation.
期刊介绍:
The Journal of Physical Organic Chemistry is the foremost international journal devoted to the relationship between molecular structure and chemical reactivity in organic systems. It publishes Research Articles, Reviews and Mini Reviews based on research striving to understand the principles governing chemical structures in relation to activity and transformation with physical and mathematical rigor, using results derived from experimental and computational methods. Physical Organic Chemistry is a central and fundamental field with multiple applications in fields such as molecular recognition, supramolecular chemistry, catalysis, photochemistry, biological and material sciences, nanotechnology and surface science.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


