{"title":"Tracking down metabolic vulnerabilities in CDK12-mutant prostate cancer","authors":"Wei-Ling Tu, Mu-En Wang, Ming Chen","doi":"10.1002/ctd2.345","DOIUrl":null,"url":null,"abstract":"<p><i>CDK12</i> is among the most frequently mutated cyclin-dependent kinases (CDKs) in various cancers, including prostate, ovarian, breast, esophageal, bladder, and colon cancers.<span><sup>1</sup></span> Specifically, biallelic aberrations of <i>CDK12</i> occur in 3%–7% of metastatic castration-resistant prostate cancer (mCRPC) cases and correlate with poor prognosis.<span><sup>1</sup></span> One of the most well-studied functions of CDK12 is its orchestration of transcription initiation and elongation through cyclin K-dependent Ser2 phosphorylation of RNA polymerase II,<span><sup>2</sup></span> and therefore CDK12 is important in regulating the expression of long genes or genes with high exon numbers, especially DNA damage response (DDR)-related genes such as <i>BRCA1</i>, <i>ATR</i>, <i>FANCI</i>, and <i>FANCD21</i>.<span><sup>3</sup></span> As a result, <i>CDK12</i>-deficient cancers are commonly characterized by focal tandem duplications (FTDs) and various features of genome instability.<span><sup>4, 5</sup></span> Because FTDs often generate a large amount of neoantigens, it has been suggested that <i>CDK12</i>-deficient tumours may be more sensitive to immunotherapy.<span><sup>5</sup></span> However, studies of immune checkpoint blockade therapy have shown only limited effects in mCRPC patients harbouring <i>CDK12</i> mutations.<span><sup>6</sup></span> Likewise, despite the observed sensitization of <i>CDK12</i>-deficient ovarian cancer cells to PARP inhibitors (PARPi) due to impaired DDR functionality,<span><sup>7</sup></span> clinical trials of PARPi have produced unsatisfactory results in prostate cancer patients with <i>CDK12</i> mutations.<span><sup>6</sup></span> Therefore, there is an urgent need to identify exploitable vulnerabilities in <i>CDK12</i>-deficient prostate cancer.</p><p>In a recent study, Zhang et al. investigated the impact of <i>CDK12</i> deficiency on cell metabolism and tumour progression in prostate cancer.<span><sup>8</sup></span> By analyzing public datasets, they confirmed an association between <i>CDK12</i> deficiency and poor prognosis in mCRPC, while noting higher levels of <i>CDK12</i> mutations in Chinese patients (15.4%–27.2%) than in the global populations (4.7%). To further delineate how <i>CDK12</i> deficiency promotes mCRPC, they generated <i>CDK12</i>-knockout prostate cancer cell lines using CRISPR-Cas9 technology and conducted metabolomic and transcriptomic analyses. The resulting data showed that <i>CDK12</i> deficiency reprogrammed energy metabolisms in prostate cancer cells; specifically, <i>CDK12</i> knockout cells exhibited higher levels of metabolites related to glycolysis, glutaminolysis, and the tricarboxylic acid cycle, but lower levels of metabolites related to β-oxidation. Further, the associated RNA-seq data showed enrichment of mitochondrial electron transport chain (ETC) and oxidative phosphorylation-related pathways in <i>CDK12</i>-deficient prostate cancers, suggesting that <i>CDK12</i> deficiency promotes mCRPC progression by enhancing mitochondrial ETC-dependent energy production (Figure 1). The Zhang team also found that <i>CDK12</i> knockout cell lines consistently showed significantly increased adenosine triphosphate (ATP) production compared to controls.</p><p>ETC activity has previously been associated with the generation of reactive oxygen species, which induce various types of cell death including ferroptosis. Ferroptosis, a form of regulated cell death driven by iron-dependent lipid peroxidation, was recently identified as an emerging therapeutic target in a range of cancers including prostate cancer.<span><sup>9</sup></span> Zhang et al. tested whether ferroptosis could be a potential therapeutic vulnerability in the context of <i>CDK12</i> deficiency. Surprisingly, they found that despite depletion of intracellular antioxidant substances such as cysteine and glutathione due to ETC upregulation, <i>CDK12</i> knockout cells were more resistant to ferroptosis inducers, including RSL3 and erastin, than control cells. Mechanistically, they found that dysregulation of lipid metabolism-related genes and alterations in metabolites involved in the phospholipid biosynthesis pathway that were typical of <i>CDK12</i>-deficient prostate cancer cells reduced their susceptibility to ferroptosis. As they probed further, they identified <i>ACSL4</i> among all lipid metabolism-related genes as the most downregulated gene correlated with ferroptosis sensitivity. ChIP assays demonstrated that the direct binding of RNA polymerase II to the <i>ACSL4</i> exon 6 sequence was significantly reduced upon <i>CDK12</i> loss in both C4-2 and PC-3 cells. The Zhang team also found that <i>ACSL4</i> mRNA stabilization fractionally contributed to <i>CDK12</i> loss-induced <i>ACSL4</i> downregulation. Though it is unclear how CDK12 regulates <i>ACSL4</i> mRNA stability, RNA modifications such as N6-methyladenosine (m6A) may be involved, as it has been previously reported that lactate, which was here found to be elevated in <i>CDK12</i> knockout prostate cancer cells, regulates METTL3-mediated <i>ACSL4</i> mRNA m6A modification and stability.<span><sup>10</sup></span> While the primary recognized function of CDK12 centers on transcription initiation and elongation, the interacting factor and the mechanisms governing how CDK12 regulates specific rather than global gene expression remain unclear. Understanding the mechanisms of how CDK12 regulates specific subsets of genes could hold significant promise for the development of novel therapies with minimal side effects.</p><p>Intriguingly, Zhang et al. found that <i>CDK12</i>-deficient prostate cancer cells, while showing a ferroptosis-resistant phenotype, were vulnerable to ETC inhibitors, specifically complex I inhibitors (Figure 1). By applying the ETC inhibitor, IACS-010759, Zhang et al. were able to show effective tumour suppression effects both in vitro and in vivo. Previous studies have shown that enzalutamide-resistant prostate cancer cells increased their dependence on mitochondrial oxidative phosphorylation compared to enzalutamide-sensitive cells, suggesting that mCRPC could be vulnerable to therapies targeting mitochondrial metabolism.<span><sup>11</sup></span> Moreover, patients with <i>CDK12</i> mutation were found to develop mCRPC more rapidly.<span><sup>12</sup></span> Collectively, these results suggest that <i>CDK12</i> deficiency promotes mCRPC progression by upregulating mitochondrial ETC activity, and that targeting mitochondrial ETC could potentially serve as a promising therapeutic strategy for managing mCRPC, particularly in those patients with <i>CDK12</i>-mutant prostate cancer.</p><p>Wei-Ling Tu, Mu-En Wang, and Ming Chen contributed to the preparation, writing, and editing of the manuscript.</p><p>The authors declared no conflict of interest.</p><p>This work was supported by NIH grants (R01 CA269211 and R01 CA266510) and an Idea Development Award from the Department of Defense Prostate Cancer Research Program (W81XWH2010185) to Ming Chen. Mu-En Wang was supported in part by an Early Investigator Research Award from the Department of Defense Prostate Cancer Research Program (W81XWH2211005).</p><p>Not applicable.</p>","PeriodicalId":72605,"journal":{"name":"Clinical and translational discovery","volume":"4 4","pages":""},"PeriodicalIF":1.9000,"publicationDate":"2024-07-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ctd2.345","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical and translational discovery","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ctd2.345","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
CDK12 is among the most frequently mutated cyclin-dependent kinases (CDKs) in various cancers, including prostate, ovarian, breast, esophageal, bladder, and colon cancers.1 Specifically, biallelic aberrations of CDK12 occur in 3%–7% of metastatic castration-resistant prostate cancer (mCRPC) cases and correlate with poor prognosis.1 One of the most well-studied functions of CDK12 is its orchestration of transcription initiation and elongation through cyclin K-dependent Ser2 phosphorylation of RNA polymerase II,2 and therefore CDK12 is important in regulating the expression of long genes or genes with high exon numbers, especially DNA damage response (DDR)-related genes such as BRCA1, ATR, FANCI, and FANCD21.3 As a result, CDK12-deficient cancers are commonly characterized by focal tandem duplications (FTDs) and various features of genome instability.4, 5 Because FTDs often generate a large amount of neoantigens, it has been suggested that CDK12-deficient tumours may be more sensitive to immunotherapy.5 However, studies of immune checkpoint blockade therapy have shown only limited effects in mCRPC patients harbouring CDK12 mutations.6 Likewise, despite the observed sensitization of CDK12-deficient ovarian cancer cells to PARP inhibitors (PARPi) due to impaired DDR functionality,7 clinical trials of PARPi have produced unsatisfactory results in prostate cancer patients with CDK12 mutations.6 Therefore, there is an urgent need to identify exploitable vulnerabilities in CDK12-deficient prostate cancer.
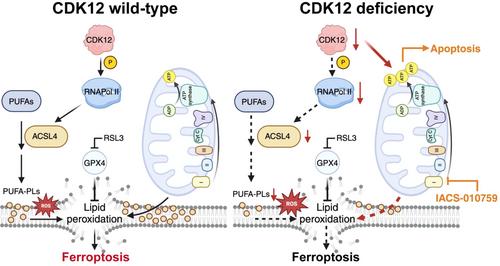
In a recent study, Zhang et al. investigated the impact of CDK12 deficiency on cell metabolism and tumour progression in prostate cancer.8 By analyzing public datasets, they confirmed an association between CDK12 deficiency and poor prognosis in mCRPC, while noting higher levels of CDK12 mutations in Chinese patients (15.4%–27.2%) than in the global populations (4.7%). To further delineate how CDK12 deficiency promotes mCRPC, they generated CDK12-knockout prostate cancer cell lines using CRISPR-Cas9 technology and conducted metabolomic and transcriptomic analyses. The resulting data showed that CDK12 deficiency reprogrammed energy metabolisms in prostate cancer cells; specifically, CDK12 knockout cells exhibited higher levels of metabolites related to glycolysis, glutaminolysis, and the tricarboxylic acid cycle, but lower levels of metabolites related to β-oxidation. Further, the associated RNA-seq data showed enrichment of mitochondrial electron transport chain (ETC) and oxidative phosphorylation-related pathways in CDK12-deficient prostate cancers, suggesting that CDK12 deficiency promotes mCRPC progression by enhancing mitochondrial ETC-dependent energy production (Figure 1). The Zhang team also found that CDK12 knockout cell lines consistently showed significantly increased adenosine triphosphate (ATP) production compared to controls.
ETC activity has previously been associated with the generation of reactive oxygen species, which induce various types of cell death including ferroptosis. Ferroptosis, a form of regulated cell death driven by iron-dependent lipid peroxidation, was recently identified as an emerging therapeutic target in a range of cancers including prostate cancer.9 Zhang et al. tested whether ferroptosis could be a potential therapeutic vulnerability in the context of CDK12 deficiency. Surprisingly, they found that despite depletion of intracellular antioxidant substances such as cysteine and glutathione due to ETC upregulation, CDK12 knockout cells were more resistant to ferroptosis inducers, including RSL3 and erastin, than control cells. Mechanistically, they found that dysregulation of lipid metabolism-related genes and alterations in metabolites involved in the phospholipid biosynthesis pathway that were typical of CDK12-deficient prostate cancer cells reduced their susceptibility to ferroptosis. As they probed further, they identified ACSL4 among all lipid metabolism-related genes as the most downregulated gene correlated with ferroptosis sensitivity. ChIP assays demonstrated that the direct binding of RNA polymerase II to the ACSL4 exon 6 sequence was significantly reduced upon CDK12 loss in both C4-2 and PC-3 cells. The Zhang team also found that ACSL4 mRNA stabilization fractionally contributed to CDK12 loss-induced ACSL4 downregulation. Though it is unclear how CDK12 regulates ACSL4 mRNA stability, RNA modifications such as N6-methyladenosine (m6A) may be involved, as it has been previously reported that lactate, which was here found to be elevated in CDK12 knockout prostate cancer cells, regulates METTL3-mediated ACSL4 mRNA m6A modification and stability.10 While the primary recognized function of CDK12 centers on transcription initiation and elongation, the interacting factor and the mechanisms governing how CDK12 regulates specific rather than global gene expression remain unclear. Understanding the mechanisms of how CDK12 regulates specific subsets of genes could hold significant promise for the development of novel therapies with minimal side effects.
Intriguingly, Zhang et al. found that CDK12-deficient prostate cancer cells, while showing a ferroptosis-resistant phenotype, were vulnerable to ETC inhibitors, specifically complex I inhibitors (Figure 1). By applying the ETC inhibitor, IACS-010759, Zhang et al. were able to show effective tumour suppression effects both in vitro and in vivo. Previous studies have shown that enzalutamide-resistant prostate cancer cells increased their dependence on mitochondrial oxidative phosphorylation compared to enzalutamide-sensitive cells, suggesting that mCRPC could be vulnerable to therapies targeting mitochondrial metabolism.11 Moreover, patients with CDK12 mutation were found to develop mCRPC more rapidly.12 Collectively, these results suggest that CDK12 deficiency promotes mCRPC progression by upregulating mitochondrial ETC activity, and that targeting mitochondrial ETC could potentially serve as a promising therapeutic strategy for managing mCRPC, particularly in those patients with CDK12-mutant prostate cancer.
Wei-Ling Tu, Mu-En Wang, and Ming Chen contributed to the preparation, writing, and editing of the manuscript.
The authors declared no conflict of interest.
This work was supported by NIH grants (R01 CA269211 and R01 CA266510) and an Idea Development Award from the Department of Defense Prostate Cancer Research Program (W81XWH2010185) to Ming Chen. Mu-En Wang was supported in part by an Early Investigator Research Award from the Department of Defense Prostate Cancer Research Program (W81XWH2211005).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


