{"title":"igRNA Prediction and Selection AI Models (igRNA-PS) for Bystander-less ABE Base Editing","authors":"","doi":"10.1016/j.jmb.2024.168714","DOIUrl":null,"url":null,"abstract":"<div><p>CRISPR derived base editing techniques tend to edit multiple bases in the targeted region, which impedes precise reversion of disease-associated single nucleotide variations (SNVs). We designed an imperfect gRNA (igRNA) editing strategy to achieve bystander-less single-base editing. To predict the performance and provide ready-to-use igRNAs, we employed a high-throughput method to edit 5000 loci, each with approximate 19 systematically designed ABE igRNAs. Through deep learning of the relationship of editing efficiency, original gRNA sequence and igRNA sequence, AI models were constructed and tested, designated igRNA Prediction and Selection AI models (igRNA-PS). The models have three functions, First, they can identify the major editing site from the bystanders on a gRNA protospacer with a near 90% accuracy. second, a modified single-base editing efficiency (SBE), considering both single-base editing efficiency and product purity, can be predicted for any given igRNAs. Third, for an editing locus, a set of 64 igRNAs derived from a gRNA can be generated, evaluated through igRNA-PS to select for the best performer, and provided to the user. In this work, we overcome one of the most significant obstacles of base editors, and provide a convenient and efficient approach for single-base bystander-less ABE base editing.</p></div>","PeriodicalId":369,"journal":{"name":"Journal of Molecular Biology","volume":null,"pages":null},"PeriodicalIF":4.7000,"publicationDate":"2024-07-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022283624003231","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
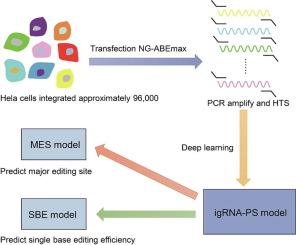
CRISPR derived base editing techniques tend to edit multiple bases in the targeted region, which impedes precise reversion of disease-associated single nucleotide variations (SNVs). We designed an imperfect gRNA (igRNA) editing strategy to achieve bystander-less single-base editing. To predict the performance and provide ready-to-use igRNAs, we employed a high-throughput method to edit 5000 loci, each with approximate 19 systematically designed ABE igRNAs. Through deep learning of the relationship of editing efficiency, original gRNA sequence and igRNA sequence, AI models were constructed and tested, designated igRNA Prediction and Selection AI models (igRNA-PS). The models have three functions, First, they can identify the major editing site from the bystanders on a gRNA protospacer with a near 90% accuracy. second, a modified single-base editing efficiency (SBE), considering both single-base editing efficiency and product purity, can be predicted for any given igRNAs. Third, for an editing locus, a set of 64 igRNAs derived from a gRNA can be generated, evaluated through igRNA-PS to select for the best performer, and provided to the user. In this work, we overcome one of the most significant obstacles of base editors, and provide a convenient and efficient approach for single-base bystander-less ABE base editing.
期刊介绍:
Journal of Molecular Biology (JMB) provides high quality, comprehensive and broad coverage in all areas of molecular biology. The journal publishes original scientific research papers that provide mechanistic and functional insights and report a significant advance to the field. The journal encourages the submission of multidisciplinary studies that use complementary experimental and computational approaches to address challenging biological questions.
Research areas include but are not limited to: Biomolecular interactions, signaling networks, systems biology; Cell cycle, cell growth, cell differentiation; Cell death, autophagy; Cell signaling and regulation; Chemical biology; Computational biology, in combination with experimental studies; DNA replication, repair, and recombination; Development, regenerative biology, mechanistic and functional studies of stem cells; Epigenetics, chromatin structure and function; Gene expression; Membrane processes, cell surface proteins and cell-cell interactions; Methodological advances, both experimental and theoretical, including databases; Microbiology, virology, and interactions with the host or environment; Microbiota mechanistic and functional studies; Nuclear organization; Post-translational modifications, proteomics; Processing and function of biologically important macromolecules and complexes; Molecular basis of disease; RNA processing, structure and functions of non-coding RNAs, transcription; Sorting, spatiotemporal organization, trafficking; Structural biology; Synthetic biology; Translation, protein folding, chaperones, protein degradation and quality control.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


