Felipe Cardoso Prado Martins , Fernanda dos Reis Rocho , Vinícius Bonatto , Pedro Henrique Jatai Batista , Jerônimo Lameira , Andrei Leitão , Carlos A. Montanari
{"title":"Novel selective proline-based peptidomimetics for human cathepsin K inhibition","authors":"Felipe Cardoso Prado Martins , Fernanda dos Reis Rocho , Vinícius Bonatto , Pedro Henrique Jatai Batista , Jerônimo Lameira , Andrei Leitão , Carlos A. Montanari","doi":"10.1016/j.bmcl.2024.129887","DOIUrl":null,"url":null,"abstract":"<div><p>Human cathepsin K (CatK) stands out as a promising target for the treatment of osteoporosis, considering its role in degrading the bone matrix. Given the small and shallow S2 subsite of CatK and considering its preference for proline or hydroxyproline, we now propose the rigidification of the leucine fragment found at the P2 position in a dipeptidyl-based inhibitor, generating rigid proline-based analogs. Accordingly, with these new proline-based peptidomimetics inhibitors, we selectively inhibited CatK against other human cathepsins (B, L and S). Among these new ligands, the most active one exhibited a high affinity (p<em>K</em><sub>i</sub> = 7.3 – 50.1 nM) for CatK and no inhibition over the other cathepsins. This specific inhibitor harbors two novel substituents never employed in other CatK inhibitors: the trifluoromethylpyrazole and the 4-methylproline at P3 and P2 positions. These results broaden and advance the path toward new potent and selective inhibitors for CatK.</p></div>","PeriodicalId":256,"journal":{"name":"Bioorganic & Medicinal Chemistry Letters","volume":null,"pages":null},"PeriodicalIF":2.5000,"publicationDate":"2024-07-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioorganic & Medicinal Chemistry Letters","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0960894X24002890","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
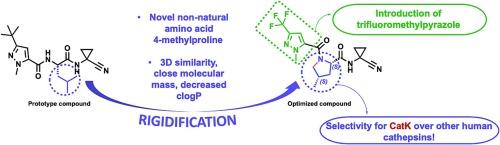
Human cathepsin K (CatK) stands out as a promising target for the treatment of osteoporosis, considering its role in degrading the bone matrix. Given the small and shallow S2 subsite of CatK and considering its preference for proline or hydroxyproline, we now propose the rigidification of the leucine fragment found at the P2 position in a dipeptidyl-based inhibitor, generating rigid proline-based analogs. Accordingly, with these new proline-based peptidomimetics inhibitors, we selectively inhibited CatK against other human cathepsins (B, L and S). Among these new ligands, the most active one exhibited a high affinity (pKi = 7.3 – 50.1 nM) for CatK and no inhibition over the other cathepsins. This specific inhibitor harbors two novel substituents never employed in other CatK inhibitors: the trifluoromethylpyrazole and the 4-methylproline at P3 and P2 positions. These results broaden and advance the path toward new potent and selective inhibitors for CatK.
期刊介绍:
Bioorganic & Medicinal Chemistry Letters presents preliminary experimental or theoretical research results of outstanding significance and timeliness on all aspects of science at the interface of chemistry and biology and on major advances in drug design and development. The journal publishes articles in the form of communications reporting experimental or theoretical results of special interest, and strives to provide maximum dissemination to a large, international audience.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


