Genomic predictions of genetic variances and correlations among traits for breeding crosses in soybean
IF 3.9
2区 生物学
Q2 ECOLOGY
引用次数: 0
Abstract
Parental selection is perhaps the most critical decision a breeder makes, establishing the foundation of the entire program for years to come. Cross selection based on predicted mean and genetic variance can be further expanded to multiple-trait improvement by predicting the genetic correlation ( $${r}_{G}$$ ) between pairs of traits. Our objective was to empirically assess the ability to predict the family mean, genetic variance, superior progeny mean and genetic correlation through genomic prediction in a soybean population. Data made available through the Soybean Nested Association Mapping project included phenotypic data on seven traits (days to maturity, lodging, oil, plant height, protein, seed size, and seed yield) for 39 families. Training population composition followed a leave-one-family-out cross-validation scheme, with the validation family genetic parameters predicted using the remaining families as the training set. The predictive abilities for family mean and superior progeny mean were significant for all traits while predictive ability of genetic variance was significant for four traits. We were able to validate significant predictive abilities of $${r}_{G}$$ for 18 out of 21 (86%) pairwise trait combinations (P < 0.05). The findings from this study support the use of genome-wide marker effects for predicting $${r}_{G}$$ in soybean biparental crosses. If successfully implemented in breeding programs, this methodology could help to increase the rate of genetic gain for multiple correlated traits.
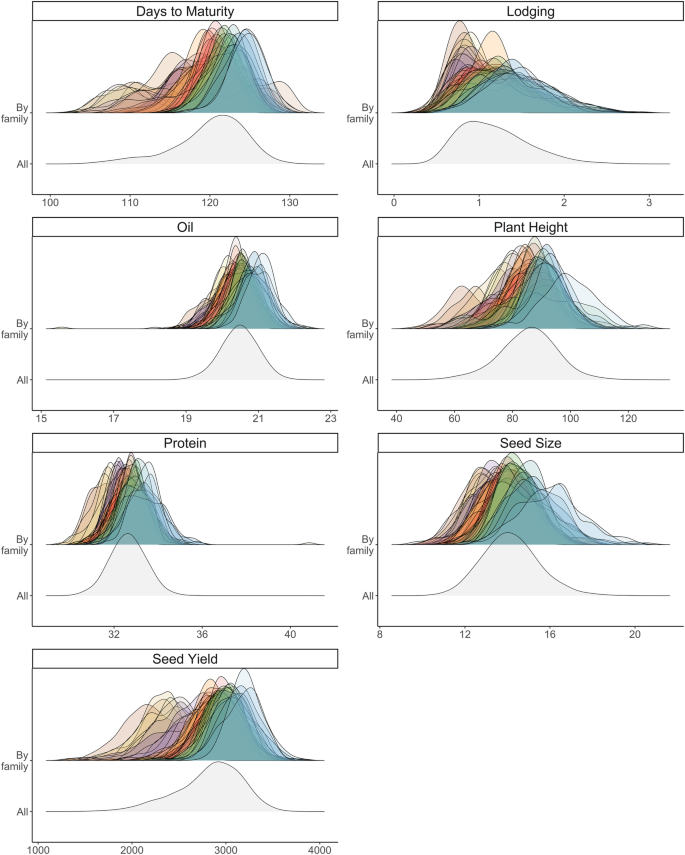
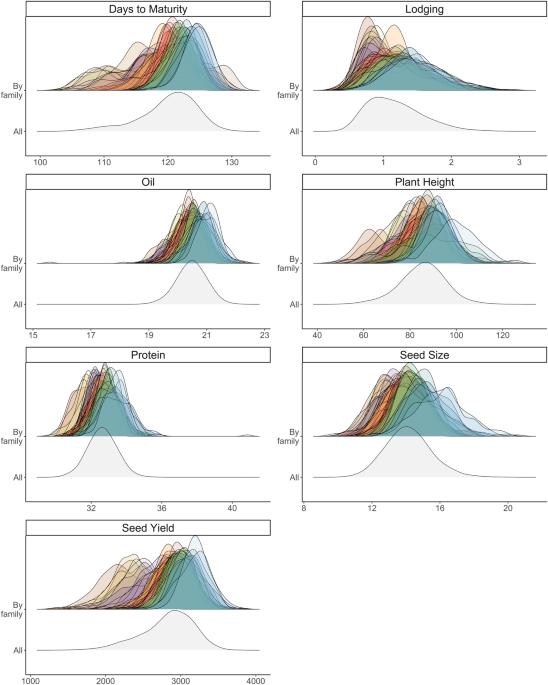
从基因组学角度预测大豆育种杂交的遗传变异和性状间的相关性。
亲本选择可能是育种者做出的最关键的决定,它奠定了整个育种计划未来几年的基础。基于预测平均值和遗传变异的杂交选育可通过预测成对性状之间的遗传相关性(r G)进一步扩展到多性状改良。我们的目标是通过大豆群体的基因组预测,对预测家系平均值、遗传变异、优良后代平均值和遗传相关性的能力进行实证评估。大豆嵌套关联图谱项目提供的数据包括 39 个家系的七个性状(成熟天数、结实率、油分、株高、蛋白质、种子大小和种子产量)的表型数据。训练群体的组成采用了 "留一族不留 "的交叉验证方案,以其余族为训练集预测验证族的遗传参数。对所有性状而言,家系平均值和优系后代平均值的预测能力都是显著的,而对四个性状而言,遗传变异的预测能力是显著的。在大豆双亲杂交的 21 个配对性状组合(P r G)中,我们验证了 18 个(86%)配对性状组合的 r G 具有显著的预测能力。如果能在育种计划中成功实施,该方法将有助于提高多个相关性状的遗传增益率。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Heredity
生物-进化生物学
CiteScore
7.50
自引率
2.60%
发文量
84
审稿时长
4-8 weeks
期刊介绍:
Heredity is the official journal of the Genetics Society. It covers a broad range of topics within the field of genetics and therefore papers must address conceptual or applied issues of interest to the journal''s wide readership
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


