{"title":"<i>PTPN11</i> and <i>FLNA</i> variants in a boy with ambiguous genitalia, short stature, and non-specific dysmorphic features.","authors":"Yuki Muranishi, Tomoyo Itonaga, Kenji Ihara, Yuko Katoh-Fukui, Satoshi Tamaoka, Atsushi Hattori, Masafumi Kon, Nobuo Shinohara, Maki Fukami","doi":"10.1297/cpe.2023-0074","DOIUrl":null,"url":null,"abstract":"<p><p>Noonan syndrome is a congenital disorder characterized by distinctive facial appearance, congenital heart defects, short stature, and skeletal dysplasia. Although boys with Noonan syndrome frequently exhibit cryptorchidism, a mild form of 46,XY disorders of sex development (DSD), they barely manifest more severe genital abnormalities. Here, we report a boy with ambiguous genitalia, short stature, and non-specific dysmorphic features. He had no cardiac abnormalities or skeletal dysplasia. His score in the Noonan syndrome diagnostic criteria (36 of 157 points, 23%) was lower than the cutoff for diagnosis (50%). Whole-exome sequencing identified a <i>de novo</i> heterozygous variant (c.922A>G: p.Asn308Asp) in <i>PTPN11</i> and a maternally inherited hemizygous variant (c.1439C>T: p.Pro480Leu) in <i>FLNA</i>. The <i>PTPN11</i> variant was a known causative mutation for Noonan syndrome. <i>FLNA</i> is a causative gene for neurodevelopmental and skeletal abnormalities and has also been implicated in 46,XY DSD. The p.Pro480Leu variant of <i>FLNA</i> was assessed as deleterious by <i>in silico</i> analyses. These results provide evidence that whole-exome sequencing is a powerful tool for diagnosing patients with atypical disease manifestations. Furthermore, our data suggest a possible role of digenic mutations as phenotypic modifiers of Noonan syndrome.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":"33 3","pages":"169-173"},"PeriodicalIF":1.2000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11234187/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2023-0074","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/5/3 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
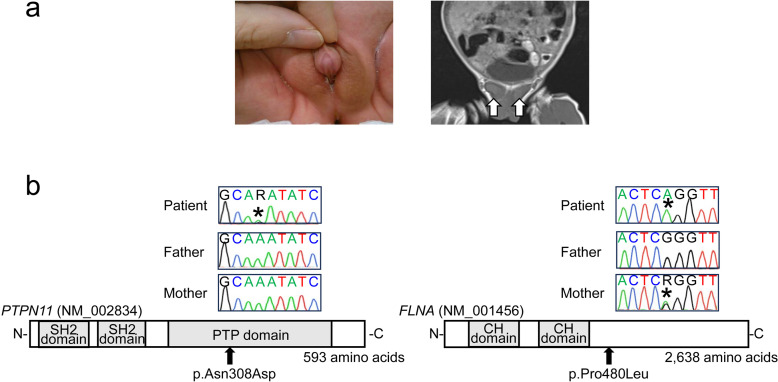
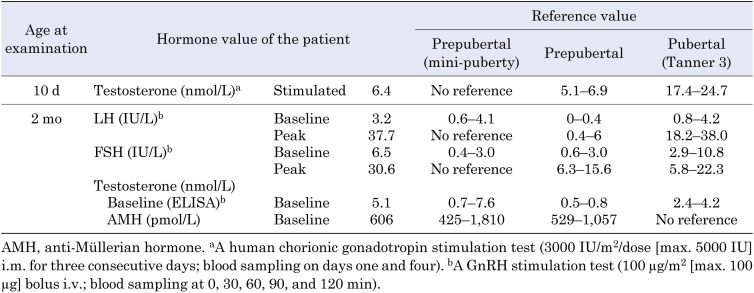
Noonan syndrome is a congenital disorder characterized by distinctive facial appearance, congenital heart defects, short stature, and skeletal dysplasia. Although boys with Noonan syndrome frequently exhibit cryptorchidism, a mild form of 46,XY disorders of sex development (DSD), they barely manifest more severe genital abnormalities. Here, we report a boy with ambiguous genitalia, short stature, and non-specific dysmorphic features. He had no cardiac abnormalities or skeletal dysplasia. His score in the Noonan syndrome diagnostic criteria (36 of 157 points, 23%) was lower than the cutoff for diagnosis (50%). Whole-exome sequencing identified a de novo heterozygous variant (c.922A>G: p.Asn308Asp) in PTPN11 and a maternally inherited hemizygous variant (c.1439C>T: p.Pro480Leu) in FLNA. The PTPN11 variant was a known causative mutation for Noonan syndrome. FLNA is a causative gene for neurodevelopmental and skeletal abnormalities and has also been implicated in 46,XY DSD. The p.Pro480Leu variant of FLNA was assessed as deleterious by in silico analyses. These results provide evidence that whole-exome sequencing is a powerful tool for diagnosing patients with atypical disease manifestations. Furthermore, our data suggest a possible role of digenic mutations as phenotypic modifiers of Noonan syndrome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


