Molecular set representation learning
IF 18.8
1区 计算机科学
Q1 COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE
引用次数: 0
Abstract
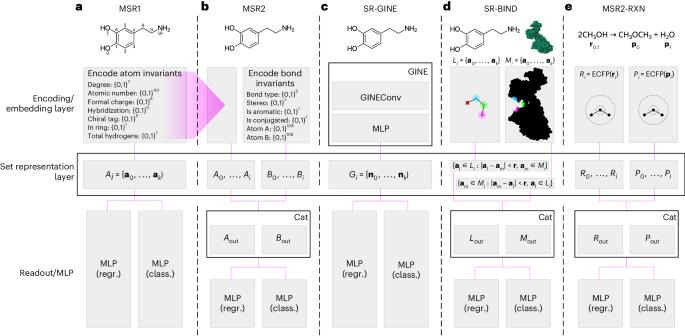
Computational representation of molecules can take many forms, including graphs, string encodings of graphs, binary vectors or learned embeddings in the form of real-valued vectors. These representations are then used in downstream classification and regression tasks using a wide range of machine learning models. However, existing models come with limitations, such as the requirement for clearly defined chemical bonds, which often do not represent the true underlying nature of a molecule. Here we propose a framework for molecular machine learning tasks based on set representation learning. We show that learning on sets of atom invariants alone reaches the performance of state-of-the-art graph-based models on the most-used chemical benchmark datasets and that introducing a set representation layer into graph neural networks can surpass the performance of established methods in the domains of chemistry, biology and material science. We introduce specialized set representation-based neural network architectures for reaction-yield and protein–ligand binding-affinity prediction. Overall, we show that the technique we denote molecular set representation learning is both an alternative and an extension to graph neural network architectures for machine learning tasks on molecules, molecule complexes and chemical reactions. Machine learning methods for molecule predictions use various representations of molecules such as in the form of strings or graphs. As an extension of graph representation learning, Probst and colleagues propose to represent a molecule as a set of atoms, to better capture the underlying chemical nature, and demonstrate improved performance in a range of machine learning tasks.

分子集表示学习
分子的计算表示有多种形式,包括图、图的字符串编码、二进制向量或实值向量形式的学习嵌入。然后,这些表征可通过各种机器学习模型用于下游分类和回归任务。然而,现有的模型有其局限性,例如需要明确定义的化学键,而这些化学键往往不能代表分子的真实基本性质。在此,我们提出了一个基于集合表示学习的分子机器学习任务框架。我们的研究表明,在最常用的化学基准数据集上,仅原子不变式集学习就能达到最先进的基于图的模型的性能,而在图神经网络中引入集合表示层,就能在化学、生物学和材料科学领域超越已有方法的性能。我们为反应产率和蛋白质配体结合亲和力预测引入了基于集合表示的专门神经网络架构。总之,我们表明,我们称之为分子集表示学习的技术既是图神经网络架构的一种替代方法,也是对分子、分子复合物和化学反应机器学习任务的一种扩展。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Machine Intelligence
Multiple-
CiteScore
36.90
自引率
2.10%
发文量
127
期刊介绍:
Nature Machine Intelligence is a distinguished publication that presents original research and reviews on various topics in machine learning, robotics, and AI. Our focus extends beyond these fields, exploring their profound impact on other scientific disciplines, as well as societal and industrial aspects. We recognize limitless possibilities wherein machine intelligence can augment human capabilities and knowledge in domains like scientific exploration, healthcare, medical diagnostics, and the creation of safe and sustainable cities, transportation, and agriculture. Simultaneously, we acknowledge the emergence of ethical, social, and legal concerns due to the rapid pace of advancements.
To foster interdisciplinary discussions on these far-reaching implications, Nature Machine Intelligence serves as a platform for dialogue facilitated through Comments, News Features, News & Views articles, and Correspondence. Our goal is to encourage a comprehensive examination of these subjects.
Similar to all Nature-branded journals, Nature Machine Intelligence operates under the guidance of a team of skilled editors. We adhere to a fair and rigorous peer-review process, ensuring high standards of copy-editing and production, swift publication, and editorial independence.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


