{"title":"A hybrid evaluation of the intestinal absorption performance of compounds from molecular structure","authors":"Mengke Sheng, Lina Ma, Zhixun Li, Xinhui Peng, Shuai Cen, Minfang Feng, Yuting Tian, Xingxing Dai, Xinyuan Shi","doi":"10.1111/cbdd.14576","DOIUrl":null,"url":null,"abstract":"<p>Intestinal absorption of compounds is significant in drug research and development. To evaluate this efficiently, a method combining mathematical modeling and molecular simulation was proposed, from the perspective of molecular structure. Based on the quantitative structure–property relationship study, the model between molecular structure and their apparent permeability coefficients was successfully constructed and verified, predicting intestinal absorption of drugs and interpreting decisive structural factors, such as AlogP98, Hydrogen bond donor and Ellipsoidal volume. The molecules with strong lipophilicity, less hydrogen bond donors and receptors, and small molecular volume are more easily absorbed. Then, the molecular dynamics simulation and molecular docking were utilized to study the mechanism of differences in intestinal absorption of drugs and investigate the role of molecular structure. Results indicated that molecules with strong lipophilicity and small volume interacted with the membrane at a lower energy and were easier to penetrate the membrane. Likewise, they had weaker interaction with P-glycoprotein and were easier to escape from it and harder to export from the body. More in, less out, is the main reason these molecules absorb well.</p>","PeriodicalId":143,"journal":{"name":"Chemical Biology & Drug Design","volume":"104 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-07-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Biology & Drug Design","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cbdd.14576","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
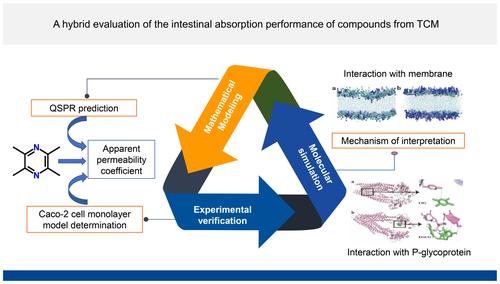
Abstract
Intestinal absorption of compounds is significant in drug research and development. To evaluate this efficiently, a method combining mathematical modeling and molecular simulation was proposed, from the perspective of molecular structure. Based on the quantitative structure–property relationship study, the model between molecular structure and their apparent permeability coefficients was successfully constructed and verified, predicting intestinal absorption of drugs and interpreting decisive structural factors, such as AlogP98, Hydrogen bond donor and Ellipsoidal volume. The molecules with strong lipophilicity, less hydrogen bond donors and receptors, and small molecular volume are more easily absorbed. Then, the molecular dynamics simulation and molecular docking were utilized to study the mechanism of differences in intestinal absorption of drugs and investigate the role of molecular structure. Results indicated that molecules with strong lipophilicity and small volume interacted with the membrane at a lower energy and were easier to penetrate the membrane. Likewise, they had weaker interaction with P-glycoprotein and were easier to escape from it and harder to export from the body. More in, less out, is the main reason these molecules absorb well.
化合物的肠道吸收在药物研发中意义重大。为了有效评估这一问题,研究人员从分子结构的角度出发,提出了一种数学建模与分子模拟相结合的方法。在定量结构-性质关系研究的基础上,成功构建并验证了分子结构与表观渗透系数之间的模型,预测了药物的肠道吸收,并解释了AlogP98、氢键供体和椭圆体体积等决定性结构因素。亲脂性强、氢键供体和受体少、分子体积小的分子更容易被吸收。然后,利用分子动力学模拟和分子对接研究了药物在肠道吸收的差异机理,并探讨了分子结构的作用。结果表明,亲脂性强、体积小的分子与膜的相互作用能量较低,更容易穿透膜。同样,它们与 P 糖蛋白的相互作用较弱,更容易从 P 糖蛋白中逃脱,也更难从体内排出。多进少出是这些分子吸收好的主要原因。
期刊介绍:
Chemical Biology & Drug Design is a peer-reviewed scientific journal that is dedicated to the advancement of innovative science, technology and medicine with a focus on the multidisciplinary fields of chemical biology and drug design. It is the aim of Chemical Biology & Drug Design to capture significant research and drug discovery that highlights new concepts, insight and new findings within the scope of chemical biology and drug design.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


