Linley Mangini, Roger Lawrence, Manuel E. Lopez, Timothy C. Graham, Christopher R. Bauer, Hang Nguyen, Cheng Su, John Ramphal, Brett E. Crawford, Tom A. Hartl
{"title":"Galactokinase 1 is the source of elevated galactose-1-phosphate and cerebrosides are modestly reduced in a mouse model of classic galactosemia","authors":"Linley Mangini, Roger Lawrence, Manuel E. Lopez, Timothy C. Graham, Christopher R. Bauer, Hang Nguyen, Cheng Su, John Ramphal, Brett E. Crawford, Tom A. Hartl","doi":"10.1002/jmd2.12438","DOIUrl":null,"url":null,"abstract":"<p>Classic galactosemia (CG) arises from loss-of-function mutations in the <i>Galt</i> gene, which codes for the enzyme galactose-1-phosphate uridylyltransferase (GALT), a central component in galactose metabolism. The neonatal fatality associated with CG can be prevented by galactose dietary restriction, but for decades it has been known that limiting galactose intake is not a cure and patients often have lasting complications. Even on a low-galactose diet, GALT's substrate galactose-1-phosphate (Gal1P) is elevated and one hypothesis is that elevated Gal1P is a driver of pathology. Here we show that Gal1P levels were elevated above wildtype (WT) in <i>Galt</i> mutant mice, while mice doubly mutant for <i>Galt</i> and the gene encoding galactokinase 1 <i>(Galk1)</i> had normal Gal1P levels. This indicates that GALK1 is necessary for the elevated Gal1P in CG. Another hypothesis to explain the pathology is that an inability to metabolize galactose leads to diminished or disrupted galactosylation of proteins or lipids. Our studies reveal that levels of a subset of cerebrosides—galactosylceramide 24:1, sulfatide 24:1, and glucosylceramide 24:1—were modestly decreased compared to WT. In contrast, gangliosides were unaltered. The observed reduction in these 24:1 cerebrosides may be relevant to the clinical pathology of CG, since the cerebroside galactosylceramide is an important structural component of myelin, the 24:1 species is the most abundant in myelin, and irregularities in white matter, of which myelin is a constituent, have been observed in patients with CG. Therefore, impaired cerebroside production may be a contributing factor to the brain damage that is a common clinical feature of the human disease.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"65 4","pages":"280-294"},"PeriodicalIF":1.8000,"publicationDate":"2024-06-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12438","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12438","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
Abstract
Classic galactosemia (CG) arises from loss-of-function mutations in the Galt gene, which codes for the enzyme galactose-1-phosphate uridylyltransferase (GALT), a central component in galactose metabolism. The neonatal fatality associated with CG can be prevented by galactose dietary restriction, but for decades it has been known that limiting galactose intake is not a cure and patients often have lasting complications. Even on a low-galactose diet, GALT's substrate galactose-1-phosphate (Gal1P) is elevated and one hypothesis is that elevated Gal1P is a driver of pathology. Here we show that Gal1P levels were elevated above wildtype (WT) in Galt mutant mice, while mice doubly mutant for Galt and the gene encoding galactokinase 1 (Galk1) had normal Gal1P levels. This indicates that GALK1 is necessary for the elevated Gal1P in CG. Another hypothesis to explain the pathology is that an inability to metabolize galactose leads to diminished or disrupted galactosylation of proteins or lipids. Our studies reveal that levels of a subset of cerebrosides—galactosylceramide 24:1, sulfatide 24:1, and glucosylceramide 24:1—were modestly decreased compared to WT. In contrast, gangliosides were unaltered. The observed reduction in these 24:1 cerebrosides may be relevant to the clinical pathology of CG, since the cerebroside galactosylceramide is an important structural component of myelin, the 24:1 species is the most abundant in myelin, and irregularities in white matter, of which myelin is a constituent, have been observed in patients with CG. Therefore, impaired cerebroside production may be a contributing factor to the brain damage that is a common clinical feature of the human disease.
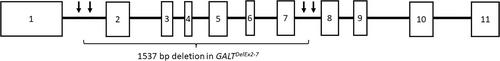

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


