{"title":"Predicting melting temperatures across the periodic table with machine learning atomistic potentials†","authors":"Christopher M. Andolina and Wissam A. Saidi","doi":"10.1039/D4DD00069B","DOIUrl":null,"url":null,"abstract":"<p >Understanding how materials melt is crucial for their practical applications and development, thereby enabling us to predict their behavior in real-world environmental conditions. Accurate computation of melting temperatures (<em>T</em><small><sub>m</sub></small>) has been a long-standing pursuit involving various methods for classical potentials and first-principles calculations. However, finding literature <em>T</em><small><sub>m</sub></small> references for many elements using a clearly defined set of calculation parameters is rare. Herein we apply deep neural network atomistic potentials (DNPs), trained on density functional theory (DFT) generated datasets, to describe the melting temperature of 20 single-element materials across the Periodic Table using large-scale molecular dynamics simulations. Our results demonstrate high-fidelity with experimental observations and also with calculated reference melting temperatures, yielding an average deviation of less than 18%. We propose a straightforward elemental-group-specific relationship between <em>T</em><small><sub>m</sub></small> and cohesive energy for these calculated references to provide reliable DFT specific reference points, which we believe can be readily applied to many materials. Additionally, we compare DNP predictions for three representative elements at external pressures up to 30 GPa in molecular dynamics simulations, revealing reasonable consistency with experimental and DFT literature references despite the lack of explicit training at these high pressures. This work further extends our flexible approach to developing and modifying DNPs to create unique atomistic potentials tailored to describe atomically complex materials under extreme environmental conditions.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":" 7","pages":" 1421-1429"},"PeriodicalIF":6.2000,"publicationDate":"2024-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d4dd00069b?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d4dd00069b","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
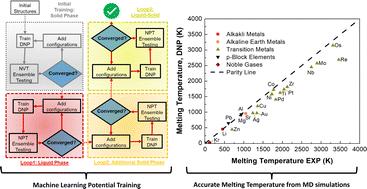
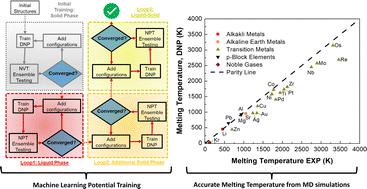
Understanding how materials melt is crucial for their practical applications and development, thereby enabling us to predict their behavior in real-world environmental conditions. Accurate computation of melting temperatures (Tm) has been a long-standing pursuit involving various methods for classical potentials and first-principles calculations. However, finding literature Tm references for many elements using a clearly defined set of calculation parameters is rare. Herein we apply deep neural network atomistic potentials (DNPs), trained on density functional theory (DFT) generated datasets, to describe the melting temperature of 20 single-element materials across the Periodic Table using large-scale molecular dynamics simulations. Our results demonstrate high-fidelity with experimental observations and also with calculated reference melting temperatures, yielding an average deviation of less than 18%. We propose a straightforward elemental-group-specific relationship between Tm and cohesive energy for these calculated references to provide reliable DFT specific reference points, which we believe can be readily applied to many materials. Additionally, we compare DNP predictions for three representative elements at external pressures up to 30 GPa in molecular dynamics simulations, revealing reasonable consistency with experimental and DFT literature references despite the lack of explicit training at these high pressures. This work further extends our flexible approach to developing and modifying DNPs to create unique atomistic potentials tailored to describe atomically complex materials under extreme environmental conditions.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


