Structure of small yttrium monoxide clusters, chemical bonding, and photoionization: threshold photoionization and density functional theory investigations†
{"title":"Structure of small yttrium monoxide clusters, chemical bonding, and photoionization: threshold photoionization and density functional theory investigations†","authors":"Varun Vinayak Deshpande, Vaibhav Chauhan, Debashis Bandyopadhyay, Anakuthil Anoop and Soumen Bhattacharyya","doi":"10.1039/D4CP02351J","DOIUrl":null,"url":null,"abstract":"<p >The photoionization (PI) spectra of small gas-phase yttrium monoxide clusters, Y<small><sub><em>n</em></sub></small>O (<em>n</em> = 1–8), are investigated, and the adiabatic ionization energies are determined. The stable structures are obtained from density functional theory (DFT) calculations. The ground state structures are further confirmed by the CCSD(T) method. The PI spectra are calculated for these stable structures and are compared with the experimental PI spectra. The ground-state structures of the neutral and cation clusters are experimentally assigned with confidence on the basis of a favourable agreement between the experimental and calculated PI spectra. New structures are proposed for Y<small><sub>2</sub></small>O, Y<small><sub>6</sub></small>O, and Y<small><sub>8</sub></small>O compared to the previous literature. Y<small><sub>2</sub></small>O is a linear molecule in the ground state that was previously proposed as a <em>C</em><small><sub>2v</sub></small> bent molecule. The Y<small><sub><em>n</em></sub></small>O clusters become 3-dimensional from <em>n</em> ≥ 3. The O atom stays outside, bridging a triangular face of yttrium clusters. Chemical bonding between the yttrium and oxygen atoms is mostly ionic. The excess charge on the oxygen atom is around 1.4<em>e</em><small><sup>−</sup></small>, transferred from the yttrium atoms bonded with it. Yttrium atoms are mostly covalently bonded. However, for the bigger clusters, free charges of both polarities appear on yttrium atoms that are not bonded with oxygen, indicating ionic interactions. Frontier orbitals consist of mainly delocalized 4d electrons with some 5s contributions, forming Y–Y bonding interactions, but with little contribution and zero contribution from the oxygen orbitals, regardless of the cluster size. The lost electron of Y<small><sub><em>n</em></sub></small>O<small><sup>+</sup></small> mostly comes from the 5s orbitals of all Y atoms in the cluster up to size <em>n</em> = 4, and then from 4d–5s hybrid orbitals from <em>n</em> ≥ 5, with the d contribution increasing with size. This is contrary to the previous view in the literature that photoionization occurs from a localized 4d orbital.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":null,"pages":null},"PeriodicalIF":2.9000,"publicationDate":"2024-07-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp02351j","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
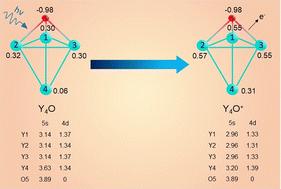
The photoionization (PI) spectra of small gas-phase yttrium monoxide clusters, YnO (n = 1–8), are investigated, and the adiabatic ionization energies are determined. The stable structures are obtained from density functional theory (DFT) calculations. The ground state structures are further confirmed by the CCSD(T) method. The PI spectra are calculated for these stable structures and are compared with the experimental PI spectra. The ground-state structures of the neutral and cation clusters are experimentally assigned with confidence on the basis of a favourable agreement between the experimental and calculated PI spectra. New structures are proposed for Y2O, Y6O, and Y8O compared to the previous literature. Y2O is a linear molecule in the ground state that was previously proposed as a C2v bent molecule. The YnO clusters become 3-dimensional from n ≥ 3. The O atom stays outside, bridging a triangular face of yttrium clusters. Chemical bonding between the yttrium and oxygen atoms is mostly ionic. The excess charge on the oxygen atom is around 1.4e−, transferred from the yttrium atoms bonded with it. Yttrium atoms are mostly covalently bonded. However, for the bigger clusters, free charges of both polarities appear on yttrium atoms that are not bonded with oxygen, indicating ionic interactions. Frontier orbitals consist of mainly delocalized 4d electrons with some 5s contributions, forming Y–Y bonding interactions, but with little contribution and zero contribution from the oxygen orbitals, regardless of the cluster size. The lost electron of YnO+ mostly comes from the 5s orbitals of all Y atoms in the cluster up to size n = 4, and then from 4d–5s hybrid orbitals from n ≥ 5, with the d contribution increasing with size. This is contrary to the previous view in the literature that photoionization occurs from a localized 4d orbital.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


