{"title":"Docking Score ML: Target-Specific Machine Learning Models Improving Docking-Based Virtual Screening in 155 Targets.","authors":"Haihan Liu, Baichun Hu, Peiying Chen, Xiao Wang, Hanxun Wang, Shizun Wang, Jian Wang, Bin Lin, Maosheng Cheng","doi":"10.1021/acs.jcim.4c00072","DOIUrl":null,"url":null,"abstract":"<p><p>In drug discovery, molecular docking methods face challenges in accurately predicting energy. Scoring functions used in molecular docking often fail to simulate complex protein-ligand interactions fully and accurately leading to biases and inaccuracies in virtual screening and target predictions. We introduce the \"Docking Score ML\", developed from an analysis of over 200,000 docked complexes from 155 known targets for cancer treatments. The scoring functions used are founded on bioactivity data sourced from ChEMBL and have been fine-tuned using both supervised machine learning and deep learning techniques. We validated our approach extensively using multiple data sets such as validation of selectivity mechanism, the DUDE, DUD-AD, and LIT-PCBA data sets, and performed a multitarget analysis on drugs like sunitinib. To enhance prediction accuracy, feature fusion techniques were explored. By merging the capabilities of the Graph Convolutional Network (GCN) with multiple docking functions, our results indicated a clear superiority of our methodologies over conventional approaches. These advantages demonstrate that Docking Score ML is an efficient and accurate tool for virtual screening and reverse docking.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"5413-5426"},"PeriodicalIF":5.3000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00072","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/3 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
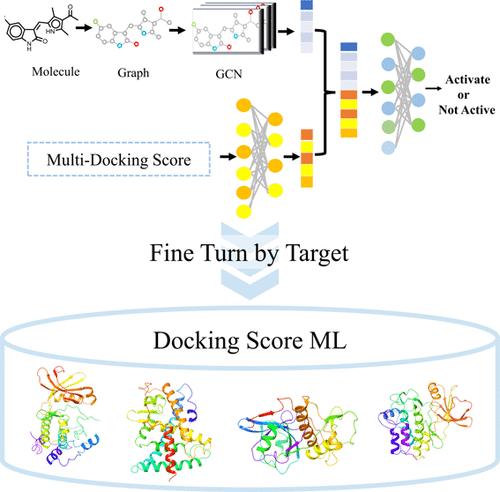
Abstract
In drug discovery, molecular docking methods face challenges in accurately predicting energy. Scoring functions used in molecular docking often fail to simulate complex protein-ligand interactions fully and accurately leading to biases and inaccuracies in virtual screening and target predictions. We introduce the "Docking Score ML", developed from an analysis of over 200,000 docked complexes from 155 known targets for cancer treatments. The scoring functions used are founded on bioactivity data sourced from ChEMBL and have been fine-tuned using both supervised machine learning and deep learning techniques. We validated our approach extensively using multiple data sets such as validation of selectivity mechanism, the DUDE, DUD-AD, and LIT-PCBA data sets, and performed a multitarget analysis on drugs like sunitinib. To enhance prediction accuracy, feature fusion techniques were explored. By merging the capabilities of the Graph Convolutional Network (GCN) with multiple docking functions, our results indicated a clear superiority of our methodologies over conventional approaches. These advantages demonstrate that Docking Score ML is an efficient and accurate tool for virtual screening and reverse docking.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


