{"title":"Developmental Causes of Focal Segmental Glomerulosclerosis.","authors":"Luna Shane Klomp, Elena Levtchenko, Rik Westland","doi":"10.1159/000538345","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Focal segmental glomerulosclerosis (FSGS) is a histological pattern of glomerular damage that includes idiopathic conditions as well as genetic and non-genetic forms. Among these various etiologies, different phenotypes within the spectrum of congenital anomalies of the kidney and urinary tract (CAKUT) have been associated with FSGS.</p><p><strong>Summary: </strong>Until recently, the main pathomechanism of how congenital kidney and urinary tract defects lead to FSGS was attributed to a reduced number of nephrons, resulting in biomechanical stress on the remaining glomeruli, detachment of podocytes, and subsequent inability to maintain normal glomerular architecture. The discovery of deleterious single-nucleotide variants in <i>PAX2</i>, a transcription factor crucial in normal kidney development and a known cause of papillorenal syndrome, in individuals with adult-onset FSGS without congenital kidney defects has shed new light on developmental defects that become evident during podocyte injury.</p><p><strong>Key message: </strong>In this mini-review, we challenge the assumption that FSGS in CAKUT is caused by glomerular hyperfiltration alone and hypothesize a multifactorial pathogenesis that includes overlapping cellular mechanisms that are activated in both damaged podocytes as well as nephron progenitor cells.</p>","PeriodicalId":73177,"journal":{"name":"Glomerular diseases","volume":"4 1","pages":"95-104"},"PeriodicalIF":0.0000,"publicationDate":"2024-03-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11216339/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Glomerular diseases","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000538345","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Focal segmental glomerulosclerosis (FSGS) is a histological pattern of glomerular damage that includes idiopathic conditions as well as genetic and non-genetic forms. Among these various etiologies, different phenotypes within the spectrum of congenital anomalies of the kidney and urinary tract (CAKUT) have been associated with FSGS.
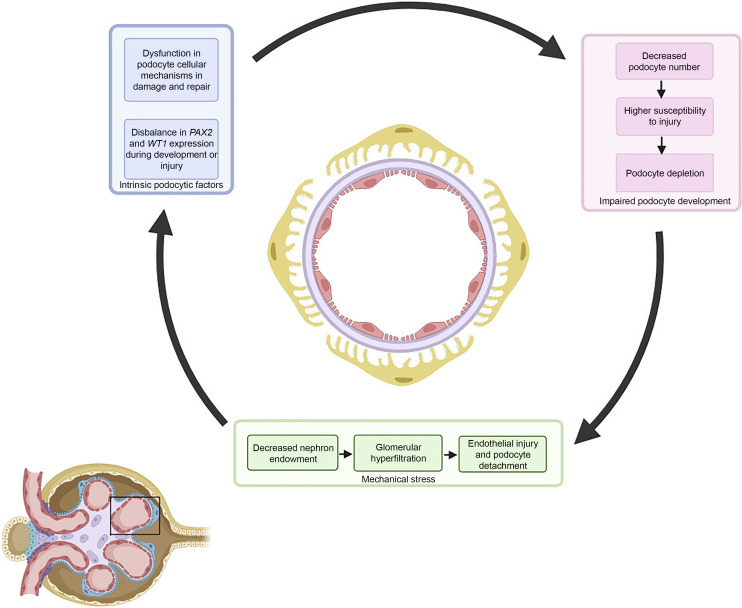
Summary: Until recently, the main pathomechanism of how congenital kidney and urinary tract defects lead to FSGS was attributed to a reduced number of nephrons, resulting in biomechanical stress on the remaining glomeruli, detachment of podocytes, and subsequent inability to maintain normal glomerular architecture. The discovery of deleterious single-nucleotide variants in PAX2, a transcription factor crucial in normal kidney development and a known cause of papillorenal syndrome, in individuals with adult-onset FSGS without congenital kidney defects has shed new light on developmental defects that become evident during podocyte injury.
Key message: In this mini-review, we challenge the assumption that FSGS in CAKUT is caused by glomerular hyperfiltration alone and hypothesize a multifactorial pathogenesis that includes overlapping cellular mechanisms that are activated in both damaged podocytes as well as nephron progenitor cells.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


