Govindan Subramanian, Kanika Manchanda, Yirong Mo, Rohit Y. Sathe, Prasad V. Bharatam
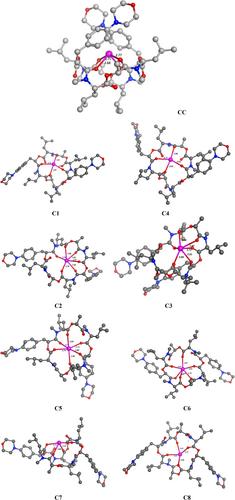
{"title":"Monovalent cation binding to model systems and the macrocyclic depsipeptide, emodepside","authors":"Govindan Subramanian, Kanika Manchanda, Yirong Mo, Rohit Y. Sathe, Prasad V. Bharatam","doi":"10.1002/jcc.27451","DOIUrl":null,"url":null,"abstract":"<p>This study focuses on the systematic exploration of the emodepside conformations bound to monovalent K<sup>+</sup> ion using quantum mechanical density functional theory (DFT) calculations at the M06-2X/6-31+G(d,p) level of theory. Nine conformers of emodepside and their complexes with K<sup>+</sup> ion were characterized as stationary points on the potential energy surface. The conformational isomers were examined for their 3D structures, bonding, energetics, and interactions with the cation. A cavitand-like structure (<b>CC</b>) is identified to be the energetically most stable arrangement. To arrive at a better understanding of the K<sup>+</sup> ion binding, calculations were initially performed on complexes formed by the K<sup>+</sup> and Na<sup>+</sup> ions with model ligands (methyl ester and N,N-dimethyl acetamide). Both the natural bond orbital (NBO) method and the block-localized wavefunction (BLW) energy decomposition approach was employed to assess the bonding and energetic contributions stabilizing the ion-bound model complexes. Finally, the solvent effect was evaluated through complete geometry optimizations and energy minimizations for the model ion-ligand complexes and the emodepside-K<sup>+</sup> bound complexes using an implicit solvent model mimicking water and DMSO.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 28","pages":"2409-2423"},"PeriodicalIF":4.8000,"publicationDate":"2024-06-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27451","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27451","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
This study focuses on the systematic exploration of the emodepside conformations bound to monovalent K+ ion using quantum mechanical density functional theory (DFT) calculations at the M06-2X/6-31+G(d,p) level of theory. Nine conformers of emodepside and their complexes with K+ ion were characterized as stationary points on the potential energy surface. The conformational isomers were examined for their 3D structures, bonding, energetics, and interactions with the cation. A cavitand-like structure (CC) is identified to be the energetically most stable arrangement. To arrive at a better understanding of the K+ ion binding, calculations were initially performed on complexes formed by the K+ and Na+ ions with model ligands (methyl ester and N,N-dimethyl acetamide). Both the natural bond orbital (NBO) method and the block-localized wavefunction (BLW) energy decomposition approach was employed to assess the bonding and energetic contributions stabilizing the ion-bound model complexes. Finally, the solvent effect was evaluated through complete geometry optimizations and energy minimizations for the model ion-ligand complexes and the emodepside-K+ bound complexes using an implicit solvent model mimicking water and DMSO.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


