Stefania Palumbo, Domenico Palumbo, Grazia Cirillo, Giorgio Giurato, Francesca Aiello, Emanuele Miraglia Del Giudice, Anna Grandone
{"title":"Methylome analysis in girls with idiopathic central precocious puberty.","authors":"Stefania Palumbo, Domenico Palumbo, Grazia Cirillo, Giorgio Giurato, Francesca Aiello, Emanuele Miraglia Del Giudice, Anna Grandone","doi":"10.1186/s13148-024-01683-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Genetic and environmental factors are implicated in many developmental processes. Recent evidence, however, has suggested that epigenetic changes may also influence the onset of puberty or the susceptibility to a wide range of diseases later in life. The present study aims to investigate changes in genomic DNA methylation profiles associated with pubertal onset analyzing human peripheral blood leukocytes from three different groups of subjects: 19 girls with central precocious puberty (CPP), 14 healthy prepubertal girls matched by age and 13 healthy pubertal girls matched by pubertal stage. For this purpose, the comparisons were performed between pre- and pubertal controls to identify changes in normal pubertal transition and CPP versus pre- and pubertal controls.</p><p><strong>Results: </strong>Analysis of methylation changes associated with normal pubertal transition identified 1006 differentially methylated CpG sites, 86% of them were found to be hypermethylated in prepubertal controls. Some of these CpG sites reside in genes associated with the age of menarche or transcription factors involved in the process of pubertal development. Analysis of methylome profiles in CPP patients showed 65% and 55% hypomethylated CpG sites compared with prepubertal and pubertal controls, respectively. In addition, interestingly, our results revealed the presence of 43 differentially methylated genes coding for zinc finger (ZNF) proteins. Gene ontology and IPA analysis performed in the three groups studied revealed significant enrichment of them in some pathways related to neuronal communication (semaphorin and gustation pathways), estrogens action, some cancers (particularly breast and ovarian) or metabolism (particularly sirtuin).</p><p><strong>Conclusions: </strong>The different methylation profiles of girls with normal and precocious puberty indicate that regulation of the pubertal process in humans is associated with specific epigenetic changes. Differentially methylated genes include ZNF genes that may play a role in developmental control. In addition, our data highlight changes in the methylation status of genes involved in signaling pathways that determine the migration and function of GnRH neurons and the onset of metabolic and neoplastic diseases that may be associated with CPP in later life.</p>","PeriodicalId":10366,"journal":{"name":"Clinical Epigenetics","volume":"16 1","pages":"82"},"PeriodicalIF":4.4000,"publicationDate":"2024-06-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11193236/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Epigenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13148-024-01683-1","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Genetic and environmental factors are implicated in many developmental processes. Recent evidence, however, has suggested that epigenetic changes may also influence the onset of puberty or the susceptibility to a wide range of diseases later in life. The present study aims to investigate changes in genomic DNA methylation profiles associated with pubertal onset analyzing human peripheral blood leukocytes from three different groups of subjects: 19 girls with central precocious puberty (CPP), 14 healthy prepubertal girls matched by age and 13 healthy pubertal girls matched by pubertal stage. For this purpose, the comparisons were performed between pre- and pubertal controls to identify changes in normal pubertal transition and CPP versus pre- and pubertal controls.
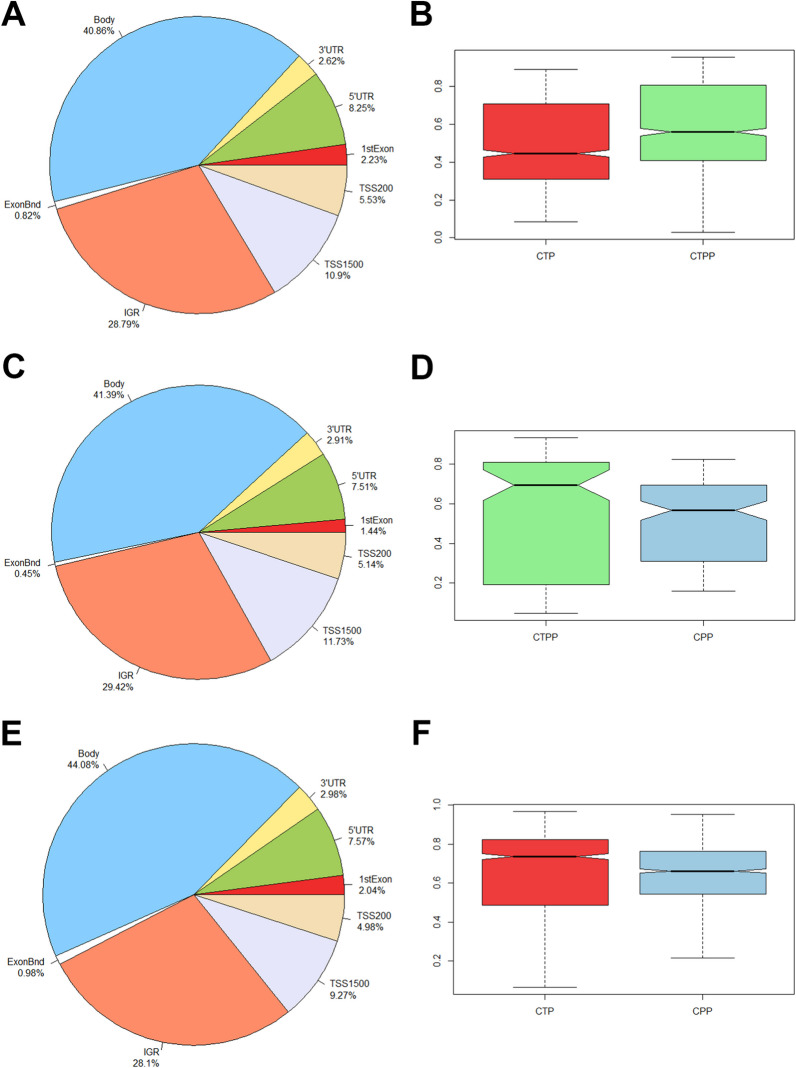
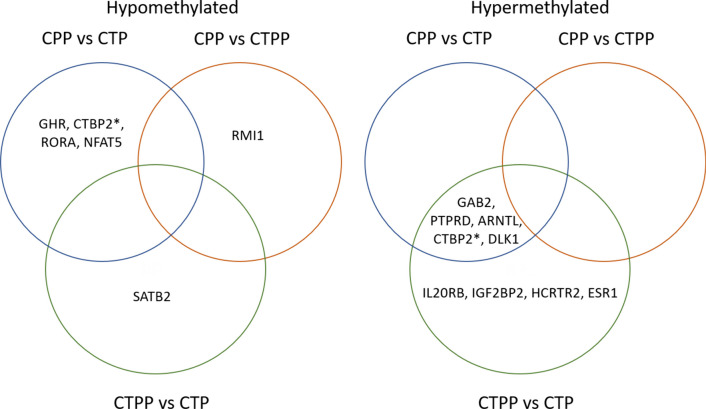
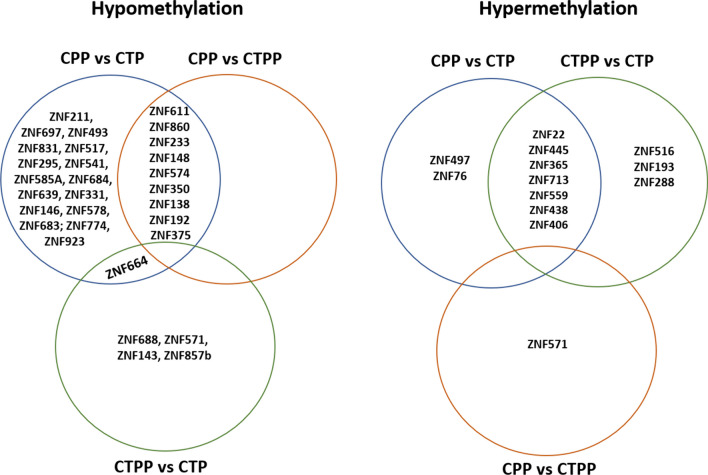
Results: Analysis of methylation changes associated with normal pubertal transition identified 1006 differentially methylated CpG sites, 86% of them were found to be hypermethylated in prepubertal controls. Some of these CpG sites reside in genes associated with the age of menarche or transcription factors involved in the process of pubertal development. Analysis of methylome profiles in CPP patients showed 65% and 55% hypomethylated CpG sites compared with prepubertal and pubertal controls, respectively. In addition, interestingly, our results revealed the presence of 43 differentially methylated genes coding for zinc finger (ZNF) proteins. Gene ontology and IPA analysis performed in the three groups studied revealed significant enrichment of them in some pathways related to neuronal communication (semaphorin and gustation pathways), estrogens action, some cancers (particularly breast and ovarian) or metabolism (particularly sirtuin).
Conclusions: The different methylation profiles of girls with normal and precocious puberty indicate that regulation of the pubertal process in humans is associated with specific epigenetic changes. Differentially methylated genes include ZNF genes that may play a role in developmental control. In addition, our data highlight changes in the methylation status of genes involved in signaling pathways that determine the migration and function of GnRH neurons and the onset of metabolic and neoplastic diseases that may be associated with CPP in later life.
期刊介绍:
Clinical Epigenetics, the official journal of the Clinical Epigenetics Society, is an open access, peer-reviewed journal that encompasses all aspects of epigenetic principles and mechanisms in relation to human disease, diagnosis and therapy. Clinical trials and research in disease model organisms are particularly welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


