{"title":"Structural, magnetic, electronic, optical and mechanical properties of actinide perovskite oxides XAnO3 [X = Cs+, Ba2+; An = Np5+, Np4+]: GGA, GGA+U and GGA+U+mBJ investigations","authors":"M. Musa Saad H.-E. , B.O. Alsobhi","doi":"10.1016/j.jmgm.2024.108815","DOIUrl":null,"url":null,"abstract":"<div><p>First-principles density functional theory (DFT)-based calculations were performed to investigate the structural, magnetic, electronic, optical and mechanical properties of two actinide perovskite oxides XAnO<sub>3</sub>, where [X = Cs<sup>+</sup>, Ba<sup>2+</sup>; An = Np<sup>5+</sup>, Np<sup>4+</sup>]. Wien2k software is utilized with GGA, GGA + U and GGA + U + mBJ potentials. The unit cell volumes for cubic (Pm-3m) structure of XAnO<sub>3</sub> are optimized to achieve the ground state energy and equilibrium parameters. Substitution of X- and An-sites increases the lattice constant, <span><math><mrow><msub><mi>a</mi><mn>0</mn></msub></mrow></math></span> = 4.3998 Å (X = Cs<sup>+</sup>) and <span><math><mrow><msub><mi>a</mi><mn>0</mn></msub></mrow></math></span> = 4.4378 Å (X = Ba<sup>2+</sup>). The calculated band structure plus total and partial density of states using these methods confirm the 100 % spin-polarization and half-metallic (HM) nature of XAnO<sub>3</sub> with <span><math><mrow><msubsup><mi>E</mi><mi>g</mi><mo>↓</mo></msubsup></mrow></math></span> = 2.731, 3.896 and 3.787 eV (X = Cs<sup>+</sup>); 3.891, 3.929 and 4.329 eV (X = Cs<sup>+</sup>). Total magnetic moment per unit cell of XAnO<sub>3</sub> is respectively <span><math><mrow><msup><mi>M</mi><mrow><mi>T</mi><mi>o</mi><mi>t</mi></mrow></msup></mrow></math></span> = 2.0 and 3.0 μ<sub>B</sub> revealing their ferromagnetic (FM) behavior with high Curie temperature (<span><math><mrow><msub><mi>T</mi><mi>C</mi></msub></mrow></math></span>) within GGA, GGA + U and GGA + U + mBJ. Mechanical and thermodynamic stability of XAnO<sub>3</sub> have been proved via the elastic parameters, sound velocity, Debye and melting temperatures, and enthalpy of formation. In addition, XAnO<sub>3</sub> show amazing optical responses include high absorption, conductivity, refractivity, and reflectivity. These investigated properties confirm that XAnO<sub>3</sub> materials have FM-HM and high optical characteristics and they perfectly suitable for many spintronics and optoelectronics applications such as sensors, storage devices and photodiodes.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"131 ","pages":"Article 108815"},"PeriodicalIF":2.7000,"publicationDate":"2024-06-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001153","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
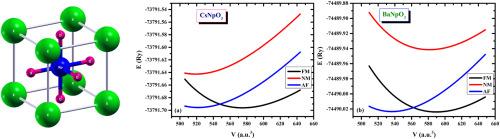
First-principles density functional theory (DFT)-based calculations were performed to investigate the structural, magnetic, electronic, optical and mechanical properties of two actinide perovskite oxides XAnO3, where [X = Cs+, Ba2+; An = Np5+, Np4+]. Wien2k software is utilized with GGA, GGA + U and GGA + U + mBJ potentials. The unit cell volumes for cubic (Pm-3m) structure of XAnO3 are optimized to achieve the ground state energy and equilibrium parameters. Substitution of X- and An-sites increases the lattice constant, = 4.3998 Å (X = Cs+) and = 4.4378 Å (X = Ba2+). The calculated band structure plus total and partial density of states using these methods confirm the 100 % spin-polarization and half-metallic (HM) nature of XAnO3 with = 2.731, 3.896 and 3.787 eV (X = Cs+); 3.891, 3.929 and 4.329 eV (X = Cs+). Total magnetic moment per unit cell of XAnO3 is respectively = 2.0 and 3.0 μB revealing their ferromagnetic (FM) behavior with high Curie temperature () within GGA, GGA + U and GGA + U + mBJ. Mechanical and thermodynamic stability of XAnO3 have been proved via the elastic parameters, sound velocity, Debye and melting temperatures, and enthalpy of formation. In addition, XAnO3 show amazing optical responses include high absorption, conductivity, refractivity, and reflectivity. These investigated properties confirm that XAnO3 materials have FM-HM and high optical characteristics and they perfectly suitable for many spintronics and optoelectronics applications such as sensors, storage devices and photodiodes.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


