Machine learning-aided generative molecular design
IF 18.8
1区 计算机科学
Q1 COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE
引用次数: 0
Abstract
Machine learning has provided a means to accelerate early-stage drug discovery by combining molecule generation and filtering steps in a single architecture that leverages the experience and design preferences of medicinal chemists. However, designing machine learning models that can achieve this on the fly to the satisfaction of medicinal chemists remains a challenge owing to the enormous search space. Researchers have addressed de novo design of molecules by decomposing the problem into a series of tasks determined by design criteria. Here we provide a comprehensive overview of the current state of the art in molecular design using machine learning models as well as important design decisions, such as the choice of molecular representations, generative methods and optimization strategies. Subsequently, we present a collection of practical applications in which the reviewed methodologies have been experimentally validated, encompassing both academic and industrial efforts. Finally, we draw attention to the theoretical, computational and empirical challenges in deploying generative machine learning and highlight future opportunities to better align such approaches to achieve realistic drug discovery end points. Data-driven generative methods have the potential to greatly facilitate molecular design tasks for drug design.
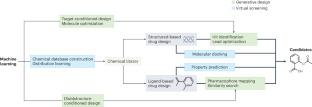
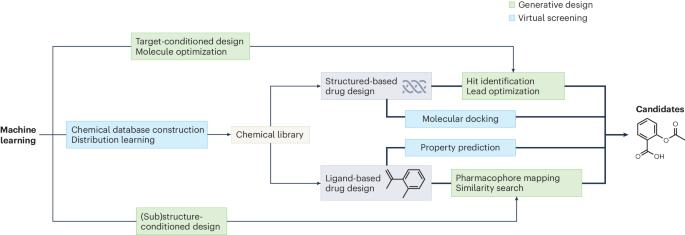
机器学习辅助生成分子设计
机器学习利用药物化学家的经验和设计偏好,将分子生成和筛选步骤结合在一个架构中,为加速早期药物发现提供了一种方法。然而,由于搜索空间巨大,要设计出能让药物化学家满意的机器学习模型仍然是一项挑战。研究人员通过将问题分解为一系列由设计标准决定的任务来解决分子的从头设计问题。在此,我们将全面概述目前使用机器学习模型进行分子设计的最新技术,以及重要的设计决策,如分子表征、生成方法和优化策略的选择。随后,我们介绍了一系列实际应用,其中所回顾的方法已在实验中得到验证,包括学术界和工业界的努力。最后,我们提请大家注意在部署生成式机器学习时所面临的理论、计算和经验方面的挑战,并强调未来有机会更好地调整这些方法,以实现现实的药物发现终点。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Machine Intelligence
Multiple-
CiteScore
36.90
自引率
2.10%
发文量
127
期刊介绍:
Nature Machine Intelligence is a distinguished publication that presents original research and reviews on various topics in machine learning, robotics, and AI. Our focus extends beyond these fields, exploring their profound impact on other scientific disciplines, as well as societal and industrial aspects. We recognize limitless possibilities wherein machine intelligence can augment human capabilities and knowledge in domains like scientific exploration, healthcare, medical diagnostics, and the creation of safe and sustainable cities, transportation, and agriculture. Simultaneously, we acknowledge the emergence of ethical, social, and legal concerns due to the rapid pace of advancements.
To foster interdisciplinary discussions on these far-reaching implications, Nature Machine Intelligence serves as a platform for dialogue facilitated through Comments, News Features, News & Views articles, and Correspondence. Our goal is to encourage a comprehensive examination of these subjects.
Similar to all Nature-branded journals, Nature Machine Intelligence operates under the guidance of a team of skilled editors. We adhere to a fair and rigorous peer-review process, ensuring high standards of copy-editing and production, swift publication, and editorial independence.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


