Yi Rong, Nanxi Li, Xuan Qiao, Lei Yang, Peng Han, Zhiyun Meng, Hui Gan, Zhuona Wu, Xiaoxia Zhu, Yunbo Sun, Shuchen Liu, Guifang Dou, Ruolan Gu
{"title":"Icaritin exhibits potential drug–drug interactions through the inhibition of human UDP-glucuronosyltransferase in vitro","authors":"Yi Rong, Nanxi Li, Xuan Qiao, Lei Yang, Peng Han, Zhiyun Meng, Hui Gan, Zhuona Wu, Xiaoxia Zhu, Yunbo Sun, Shuchen Liu, Guifang Dou, Ruolan Gu","doi":"10.1002/bdd.2397","DOIUrl":null,"url":null,"abstract":"<p>Icaritin is a prenylflavonoid derivative of the genus Epimedium (Berberidaceae) and has a variety of pharmacological actions. Icaritin is approved by the National Medical Products Administration as an anticancer drug that exhibits efficacy and safety advantages in patients with hepatocellular carcinoma cells. This study aimed to evaluate the inhibitory effects of icaritin on UDP-glucuronosyltransferase (UGT) isoforms. 4-Methylumbelliferone (4-MU) was employed as a probe drug for all the tested UGT isoforms using in vitro human liver microsomes (HLM). The inhibition potentials of UGT1A1 and 1A9 in HLM were further tested by employing 17β-estradiol (E2) and propofol (PRO) as probe substrates, respectively. The results showed that icaritin inhibits UGT1A1, 1A3, 1A4, 1A7, 1A8, 1A10, 2B7, and 2B15. Furthermore, icaritin exhibited a mixed inhibition of UGT1A1, 1A3, and 1A9, and the inhibition kinetic parameters (K<sub>i</sub>) were calculated to be 3.538, 2.117, and 0.306 (μM), respectively. The inhibition of human liver microsomal UGT1A1 and 1A9 both followed mixed mechanism, with K<sub>i</sub> values of 2.694 and 1.431 (μM). This study provides supporting information for understanding the drug–drug interaction (DDI) potential of the flavonoid icaritin and other UGT-metabolized drugs in clinical settings. In addition, the findings provide safety evidence for DDI when liver cancer patients receive a combination therapy including icaritin.</p>","PeriodicalId":8865,"journal":{"name":"Biopharmaceutics & Drug Disposition","volume":"45 3","pages":"149-158"},"PeriodicalIF":2.0000,"publicationDate":"2024-06-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biopharmaceutics & Drug Disposition","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/bdd.2397","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
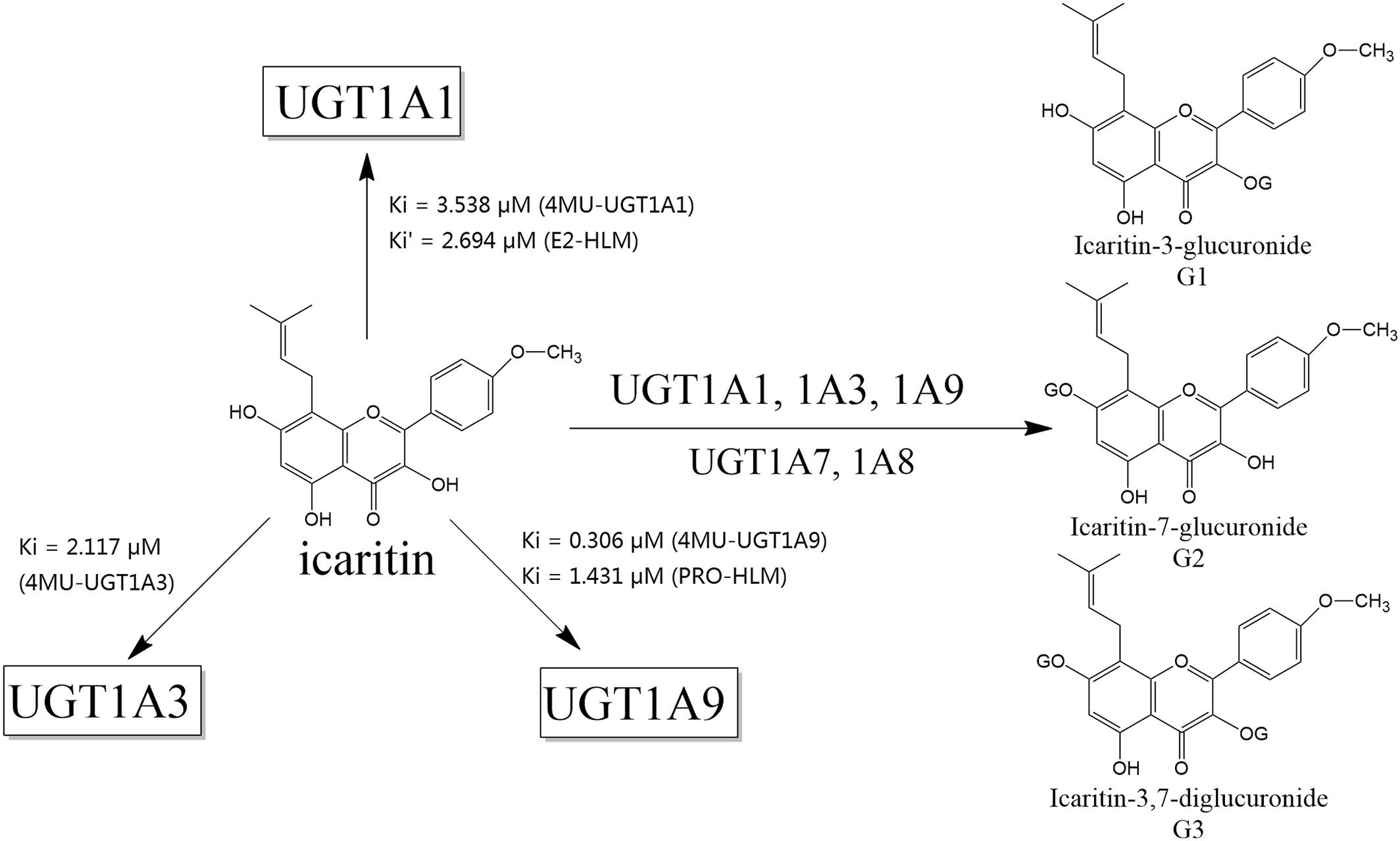
Icaritin is a prenylflavonoid derivative of the genus Epimedium (Berberidaceae) and has a variety of pharmacological actions. Icaritin is approved by the National Medical Products Administration as an anticancer drug that exhibits efficacy and safety advantages in patients with hepatocellular carcinoma cells. This study aimed to evaluate the inhibitory effects of icaritin on UDP-glucuronosyltransferase (UGT) isoforms. 4-Methylumbelliferone (4-MU) was employed as a probe drug for all the tested UGT isoforms using in vitro human liver microsomes (HLM). The inhibition potentials of UGT1A1 and 1A9 in HLM were further tested by employing 17β-estradiol (E2) and propofol (PRO) as probe substrates, respectively. The results showed that icaritin inhibits UGT1A1, 1A3, 1A4, 1A7, 1A8, 1A10, 2B7, and 2B15. Furthermore, icaritin exhibited a mixed inhibition of UGT1A1, 1A3, and 1A9, and the inhibition kinetic parameters (Ki) were calculated to be 3.538, 2.117, and 0.306 (μM), respectively. The inhibition of human liver microsomal UGT1A1 and 1A9 both followed mixed mechanism, with Ki values of 2.694 and 1.431 (μM). This study provides supporting information for understanding the drug–drug interaction (DDI) potential of the flavonoid icaritin and other UGT-metabolized drugs in clinical settings. In addition, the findings provide safety evidence for DDI when liver cancer patients receive a combination therapy including icaritin.
期刊介绍:
Biopharmaceutics & Drug Dispositionpublishes original review articles, short communications, and reports in biopharmaceutics, drug disposition, pharmacokinetics and pharmacodynamics, especially those that have a direct relation to the drug discovery/development and the therapeutic use of drugs. These includes:
- animal and human pharmacological studies that focus on therapeutic response. pharmacodynamics, and toxicity related to plasma and tissue concentrations of drugs and their metabolites,
- in vitro and in vivo drug absorption, distribution, metabolism, transport, and excretion studies that facilitate investigations related to the use of drugs in man
- studies on membrane transport and enzymes, including their regulation and the impact of pharmacogenomics on drug absorption and disposition,
- simulation and modeling in drug discovery and development
- theoretical treatises
- includes themed issues and reviews
and exclude manuscripts on
- bioavailability studies reporting only on simple PK parameters such as Cmax, tmax and t1/2 without mechanistic interpretation
- analytical methods

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


