S. Ferahtia , S. Benyettou , S. Saib , N. Bouarissa , Kh Ouail
{"title":"Mechanical and thermoelectric properties of ZrX2 and HfX2 (X = S and Se) from Van der Waals density-functional theory","authors":"S. Ferahtia , S. Benyettou , S. Saib , N. Bouarissa , Kh Ouail","doi":"10.1016/j.jmgm.2024.108812","DOIUrl":null,"url":null,"abstract":"<div><p>The structural, mechanical, and thermoelectric characteristics of layered transition metal dichalcogenides MX<sub>2</sub> (M = Zr, Hf; X = S, Se) have been studied using density functional theory along with van der Waals correction. The exchange-correlation functional, enhanced with corrections for van der Waals interactions, has been evaluated for the hexagonal bulk structures of these materials. The analysis of elastic properties reveals that these compounds exhibit brittleness at zero pressure and conform to Born's criteria for mechanical stability. Examination of elastic constants and moduli suggests that the compounds possess reasonable machinability, moderate hardness, and anisotropy in terms of sound velocity. Transport properties, including the Seebeck coefficient, electrical conductivity, thermal conductivity, and power factor, have been computed using the semi-classical Boltzmann theory implemented in the BoltzTraP code. All investigated compounds exhibit excellent thermoelectric performance at high temperatures. This result suggests that our compounds are highly promising candidate for practical utilization in the thermoelectric scope.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"131 ","pages":"Article 108812"},"PeriodicalIF":2.7000,"publicationDate":"2024-06-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001128","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
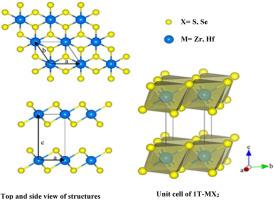
The structural, mechanical, and thermoelectric characteristics of layered transition metal dichalcogenides MX2 (M = Zr, Hf; X = S, Se) have been studied using density functional theory along with van der Waals correction. The exchange-correlation functional, enhanced with corrections for van der Waals interactions, has been evaluated for the hexagonal bulk structures of these materials. The analysis of elastic properties reveals that these compounds exhibit brittleness at zero pressure and conform to Born's criteria for mechanical stability. Examination of elastic constants and moduli suggests that the compounds possess reasonable machinability, moderate hardness, and anisotropy in terms of sound velocity. Transport properties, including the Seebeck coefficient, electrical conductivity, thermal conductivity, and power factor, have been computed using the semi-classical Boltzmann theory implemented in the BoltzTraP code. All investigated compounds exhibit excellent thermoelectric performance at high temperatures. This result suggests that our compounds are highly promising candidate for practical utilization in the thermoelectric scope.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


