Physicochemical graph neural network for learning protein–ligand interaction fingerprints from sequence data
IF 18.8
1区 计算机科学
Q1 COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE
引用次数: 0
Abstract
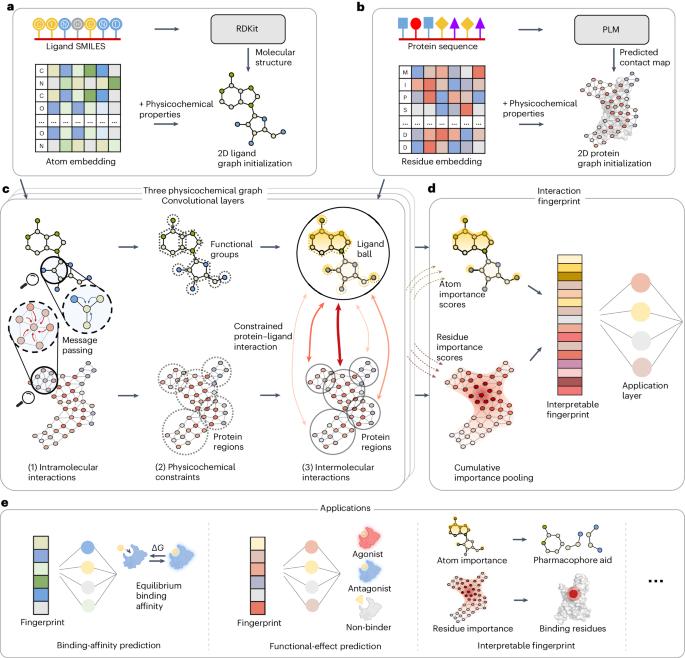
In drug discovery, determining the binding affinity and functional effects of small-molecule ligands on proteins is critical. Current computational methods can predict these protein–ligand interaction properties but often lose accuracy without high-resolution protein structures and falter in predicting functional effects. Here we introduce PSICHIC (PhySIcoCHemICal graph neural network), a framework incorporating physicochemical constraints to decode interaction fingerprints directly from sequence data alone. This enables PSICHIC to attain capabilities in decoding mechanisms underlying protein–ligand interactions, achieving state-of-the-art accuracy and interpretability. Trained on identical protein–ligand pairs without structural data, PSICHIC matched and even surpassed leading structure-based methods in binding-affinity prediction. In an experimental library screening for adenosine A1 receptor agonists, PSICHIC discerned functional effects effectively, ranking the sole novel agonist within the top three. PSICHIC’s interpretable fingerprints identified protein residues and ligand atoms involved in interactions, and helped in unveiling selectivity determinants of protein–ligand interaction. We foresee PSICHIC reshaping virtual screening and deepening our understanding of protein–ligand interactions. Predicting the binding affinity between small-molecule ligands and proteins is a key task in drug discovery; however, sequence-based methods are often less accurate than structure-based ones. Koh et al. develop a graph neural network using physicochemical constraints that discovers interactions between small molecules and proteins directly from sequence data and that can achieve state-of-the-art performance without the need for costly, experimental 3D structures.
从序列数据中学习蛋白质配体相互作用指纹的物理化学图神经网络
在药物发现过程中,确定小分子配体与蛋白质的结合亲和力和功能效应至关重要。目前的计算方法可以预测这些蛋白质-配体相互作用的特性,但如果没有高分辨率的蛋白质结构,往往会失去准确性,在预测功能效应方面也会出现偏差。在这里,我们引入了 PSICHIC(PhySIcoCHemICal graph neural network),这是一个结合了物理化学约束的框架,可直接从序列数据中解码相互作用指纹。这使得 PSICHIC 能够解码蛋白质-配体相互作用的基本机制,达到最先进的准确性和可解释性。在没有结构数据的情况下,对相同的蛋白质配体对进行训练,PSICHIC 在结合亲和力预测方面达到甚至超过了基于结构的主要方法。在腺苷 A1 受体激动剂的实验库筛选中,PSICHIC 有效地识别了功能效应,将唯一的新型激动剂排在了前三位。PSICHIC 可解释的指纹识别出了参与相互作用的蛋白质残基和配体原子,并帮助揭示了蛋白质-配体相互作用的选择性决定因素。我们预计 PSICHIC 将重塑虚拟筛选,加深我们对蛋白质配体相互作用的理解。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Machine Intelligence
Multiple-
CiteScore
36.90
自引率
2.10%
发文量
127
期刊介绍:
Nature Machine Intelligence is a distinguished publication that presents original research and reviews on various topics in machine learning, robotics, and AI. Our focus extends beyond these fields, exploring their profound impact on other scientific disciplines, as well as societal and industrial aspects. We recognize limitless possibilities wherein machine intelligence can augment human capabilities and knowledge in domains like scientific exploration, healthcare, medical diagnostics, and the creation of safe and sustainable cities, transportation, and agriculture. Simultaneously, we acknowledge the emergence of ethical, social, and legal concerns due to the rapid pace of advancements.
To foster interdisciplinary discussions on these far-reaching implications, Nature Machine Intelligence serves as a platform for dialogue facilitated through Comments, News Features, News & Views articles, and Correspondence. Our goal is to encourage a comprehensive examination of these subjects.
Similar to all Nature-branded journals, Nature Machine Intelligence operates under the guidance of a team of skilled editors. We adhere to a fair and rigorous peer-review process, ensuring high standards of copy-editing and production, swift publication, and editorial independence.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


