Majlen A Dilweg, Marina Gorostiola González, Martijn D de Ruiter, Nadine J Meijboom, Jacobus P D van Veldhoven, Rongfang Liu, Willem Jespers, Gerard J P van Westen, Laura H Heitman, Adriaan P IJzerman, Daan van der Es
{"title":"Exploring novel dilazep derivatives as hENT1 inhibitors and potentially covalent molecular tools.","authors":"Majlen A Dilweg, Marina Gorostiola González, Martijn D de Ruiter, Nadine J Meijboom, Jacobus P D van Veldhoven, Rongfang Liu, Willem Jespers, Gerard J P van Westen, Laura H Heitman, Adriaan P IJzerman, Daan van der Es","doi":"10.1007/s11302-024-10026-x","DOIUrl":null,"url":null,"abstract":"<p><p>The human equilibrative nucleoside transporter 1 (SLC29A1, hENT1) is a solute carrier that modulates the passive transport of nucleosides and nucleobases, such as adenosine. This nucleoside regulates various physiological processes, such as vasodilation and -constriction, neurotransmission and immune defense. Marketed drugs such as dilazep and dipyridamole have proven useful in cardiovascular afflictions, but the application of hENT1 inhibitors can be beneficial in a number of other diseases. In this study, 39 derivatives of dilazep's close analogue ST7092 were designed, synthesized and subsequently assessed using [<sup>3</sup>H]NBTI displacement assays and molecular docking. Different substitution patterns of the trimethoxy benzoates of ST7092 reduced interactions within the binding pocket, resulting in diminished hENT1 affinity. Conversely, [<sup>3</sup>H]NBTI displacement by potentially covalent compounds 14b, 14c, and 14d resulted in high affinities (K<sub>i</sub> values between 1.1 and 17.5 nM) for the transporter, primarily by the ability of accommodating the inhibitors in various ways in the binding pocket. However, any indication of covalent binding with amino acid residue C439 remained absent, conceivably as a result of decreased nucleophilic residue reactivity. In conclusion, this research introduces novel dilazep derivatives that are active as hENT1 inhibitors, along with the first high affinity dilazep derivatives equipped with an electrophilic warhead. These findings will aid the rational and structure-based development of novel hENT1 inhibitors and pharmacological tools to study hENT1's function, binding mechanisms, and its relevance in (patho)physiological conditions.</p>","PeriodicalId":20952,"journal":{"name":"Purinergic Signalling","volume":" ","pages":"289-316"},"PeriodicalIF":2.4000,"publicationDate":"2025-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12061832/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Purinergic Signalling","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11302-024-10026-x","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/15 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
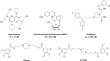
The human equilibrative nucleoside transporter 1 (SLC29A1, hENT1) is a solute carrier that modulates the passive transport of nucleosides and nucleobases, such as adenosine. This nucleoside regulates various physiological processes, such as vasodilation and -constriction, neurotransmission and immune defense. Marketed drugs such as dilazep and dipyridamole have proven useful in cardiovascular afflictions, but the application of hENT1 inhibitors can be beneficial in a number of other diseases. In this study, 39 derivatives of dilazep's close analogue ST7092 were designed, synthesized and subsequently assessed using [3H]NBTI displacement assays and molecular docking. Different substitution patterns of the trimethoxy benzoates of ST7092 reduced interactions within the binding pocket, resulting in diminished hENT1 affinity. Conversely, [3H]NBTI displacement by potentially covalent compounds 14b, 14c, and 14d resulted in high affinities (Ki values between 1.1 and 17.5 nM) for the transporter, primarily by the ability of accommodating the inhibitors in various ways in the binding pocket. However, any indication of covalent binding with amino acid residue C439 remained absent, conceivably as a result of decreased nucleophilic residue reactivity. In conclusion, this research introduces novel dilazep derivatives that are active as hENT1 inhibitors, along with the first high affinity dilazep derivatives equipped with an electrophilic warhead. These findings will aid the rational and structure-based development of novel hENT1 inhibitors and pharmacological tools to study hENT1's function, binding mechanisms, and its relevance in (patho)physiological conditions.
期刊介绍:
Nucleotides and nucleosides are primitive biological molecules that were utilized early in evolution both as intracellular energy sources and as extracellular signalling molecules. ATP was first identified as a neurotransmitter and later as a co-transmitter with all the established neurotransmitters in both peripheral and central nervous systems. Four subtypes of P1 (adenosine) receptors, 7 subtypes of P2X ion channel receptors and 8 subtypes of P2Y G protein-coupled receptors have currently been identified. Since P2 receptors were first cloned in the early 1990’s, there is clear evidence for the widespread distribution of both P1 and P2 receptor subtypes in neuronal and non-neuronal cells, including glial, immune, bone, muscle, endothelial, epithelial and endocrine cells.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


