Jenni Viitaharju, Lauri Polari, Otto Kauko, Johannes Merilahti, Anne Rokka, Diana M. Toivola, Kirsi Laitinen
{"title":"Improved breast milk proteome coverage by DIA based LC-MS/MS method","authors":"Jenni Viitaharju, Lauri Polari, Otto Kauko, Johannes Merilahti, Anne Rokka, Diana M. Toivola, Kirsi Laitinen","doi":"10.1002/pmic.202300340","DOIUrl":null,"url":null,"abstract":"<p>The breast milk composition includes a multitude of bioactive factors such as viable cells, lipids and proteins. Measuring the levels of specific proteins in breast milk plasma can be challenging because of the large dynamic range of protein concentrations and the presence of interfering substances. Therefore, most proteomic studies of breast milk have been able to identify under 1000 proteins. Optimised procedures and the latest separation technologies used in milk proteome research could lead to more precise knowledge of breast milk proteome. This study (<i>n</i> = 53) utilizes three different protein quantification methods, including direct DIA, library-based DIA method and a hybrid method combining direct DIA and library-based DIA. On average we identified 2400 proteins by hybrid method. By applying these methods, we quantified body mass index (BMI) associated variation in breast milk proteomes. There were 210 significantly different proteins when comparing the breast milk proteome of obese and overweight mothers. In addition, we analysed a small cohort (<i>n</i> = 5, randomly selected from 53 samples) by high field asymmetric waveform ion mobility spectrometry (FAIMS). FAIMS coupled with the Orbitrap Fusion Lumos mass spectrometer, which led to 41.7% higher number of protein identifications compared to Q Exactive HF mass spectrometer.</p>","PeriodicalId":224,"journal":{"name":"Proteomics","volume":"24 14","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-06-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/pmic.202300340","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomics","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/pmic.202300340","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
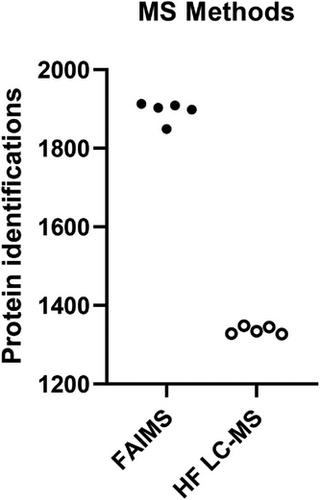
The breast milk composition includes a multitude of bioactive factors such as viable cells, lipids and proteins. Measuring the levels of specific proteins in breast milk plasma can be challenging because of the large dynamic range of protein concentrations and the presence of interfering substances. Therefore, most proteomic studies of breast milk have been able to identify under 1000 proteins. Optimised procedures and the latest separation technologies used in milk proteome research could lead to more precise knowledge of breast milk proteome. This study (n = 53) utilizes three different protein quantification methods, including direct DIA, library-based DIA method and a hybrid method combining direct DIA and library-based DIA. On average we identified 2400 proteins by hybrid method. By applying these methods, we quantified body mass index (BMI) associated variation in breast milk proteomes. There were 210 significantly different proteins when comparing the breast milk proteome of obese and overweight mothers. In addition, we analysed a small cohort (n = 5, randomly selected from 53 samples) by high field asymmetric waveform ion mobility spectrometry (FAIMS). FAIMS coupled with the Orbitrap Fusion Lumos mass spectrometer, which led to 41.7% higher number of protein identifications compared to Q Exactive HF mass spectrometer.
期刊介绍:
PROTEOMICS is the premier international source for information on all aspects of applications and technologies, including software, in proteomics and other "omics". The journal includes but is not limited to proteomics, genomics, transcriptomics, metabolomics and lipidomics, and systems biology approaches. Papers describing novel applications of proteomics and integration of multi-omics data and approaches are especially welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


