D. Christopher Braddock, Siwoo Lee and Henry S. Rzepa
{"title":"Modelling kinetic isotope effects for Swern oxidation using DFT-based transition state theory†","authors":"D. Christopher Braddock, Siwoo Lee and Henry S. Rzepa","doi":"10.1039/D3DD00246B","DOIUrl":null,"url":null,"abstract":"<p >We investigate the model reported by Giagou and Meyer in 2010 for comparing deuterium kinetic isotope effects (KIEs) computed using density functional theory (DFT) for the intramolecular hydrogen transfer step in the mechanism of the Swern oxidation of alcohols to aldehydes, with those measured by experiment. Whereas the replication of the original computed values for the gas-phase reaction proved entirely successful, several issues were discovered when a continuum solvent model was used. These included uncertainty regarding the parameters and methods used for the calculations and also the coordinates for the original reactant and transition structures, <em>via</em> their provision as data in the ESI. The original conclusions, in which a numerical mis-match between the magnitude of the computed and experimentally measured KIE was attributed to significant deviations from transition structure theory, are here instead rationalised as a manifestation of basis-set effects in the computation. Transition state theory appears to be operating successfully. We now recommend the use of basis sets of triple- or quadruple-ζ quality, rather than the split-valence level previously employed, that dispersion energy corrections be included and that a continuum solvent model using smoothed reaction cavities is essential for effective geometry optimisation and hence accurate normal coordinate analysis. An outlying experimental KIE obtained for chloroform as solvent is attributed to a small level of an explicit hydrogen bonded interaction with the substrate. A temperature outlier for the measured KIE at 195 K is suggested for further experimental investigation, although it may also be an indication of an unusually abrupt incursion of hydrogen tunnelling which would need non-Born–Oppenheimer methods in which nuclear quantum effects are included to be more accurately modelled. We predict KIEs for new substituents, of which those for R = NMe<small><sub>2</sub></small> are significantly larger than those for R = H. This approach could be useful in designing variations of the Swern reagent that could lead to synthesis of aldehydes incorporating much higher levels of deuterium. The use of FAIR data rather than the traditional model of its inclusion in the ESI is discussed, and two data discovery tools exploiting these FAIR attributes are suggested.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":" 8","pages":" 1496-1508"},"PeriodicalIF":6.2000,"publicationDate":"2024-06-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d3dd00246b?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d3dd00246b","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
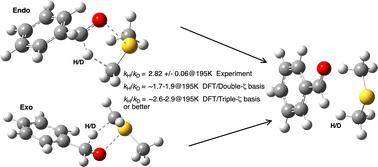
We investigate the model reported by Giagou and Meyer in 2010 for comparing deuterium kinetic isotope effects (KIEs) computed using density functional theory (DFT) for the intramolecular hydrogen transfer step in the mechanism of the Swern oxidation of alcohols to aldehydes, with those measured by experiment. Whereas the replication of the original computed values for the gas-phase reaction proved entirely successful, several issues were discovered when a continuum solvent model was used. These included uncertainty regarding the parameters and methods used for the calculations and also the coordinates for the original reactant and transition structures, via their provision as data in the ESI. The original conclusions, in which a numerical mis-match between the magnitude of the computed and experimentally measured KIE was attributed to significant deviations from transition structure theory, are here instead rationalised as a manifestation of basis-set effects in the computation. Transition state theory appears to be operating successfully. We now recommend the use of basis sets of triple- or quadruple-ζ quality, rather than the split-valence level previously employed, that dispersion energy corrections be included and that a continuum solvent model using smoothed reaction cavities is essential for effective geometry optimisation and hence accurate normal coordinate analysis. An outlying experimental KIE obtained for chloroform as solvent is attributed to a small level of an explicit hydrogen bonded interaction with the substrate. A temperature outlier for the measured KIE at 195 K is suggested for further experimental investigation, although it may also be an indication of an unusually abrupt incursion of hydrogen tunnelling which would need non-Born–Oppenheimer methods in which nuclear quantum effects are included to be more accurately modelled. We predict KIEs for new substituents, of which those for R = NMe2 are significantly larger than those for R = H. This approach could be useful in designing variations of the Swern reagent that could lead to synthesis of aldehydes incorporating much higher levels of deuterium. The use of FAIR data rather than the traditional model of its inclusion in the ESI is discussed, and two data discovery tools exploiting these FAIR attributes are suggested.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


