Maximilian G. Schuh, Davide Boldini* and Stephan A. Sieber*,
{"title":"Synergizing Chemical Structures and Bioassay Descriptions for Enhanced Molecular Property Prediction in Drug Discovery","authors":"Maximilian G. Schuh, Davide Boldini* and Stephan A. Sieber*, ","doi":"10.1021/acs.jcim.4c00765","DOIUrl":null,"url":null,"abstract":"<p >The precise prediction of molecular properties can greatly accelerate the development of new drugs. However, <i>in silico</i> molecular property prediction approaches have been limited so far to assays for which large amounts of data are available. In this study, we develop a new computational approach leveraging both the textual description of the assay of interest and the chemical structure of target compounds. By combining these two sources of information via self-supervised learning, our tool can provide accurate predictions for assays where no measurements are available. Remarkably, our approach achieves state-of-the-art performance on the FS-Mol benchmark for zero-shot prediction, outperforming a wide variety of deep learning approaches. Additionally, we demonstrate how our tool can be used for tailoring screening libraries for the assay of interest, showing promising performance in a retrospective case study on a high-throughput screening campaign. By accelerating the early identification of active molecules in drug discovery and development, this method has the potential to streamline the identification of novel therapeutics.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":null,"pages":null},"PeriodicalIF":5.6000,"publicationDate":"2024-06-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11200265/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00765","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
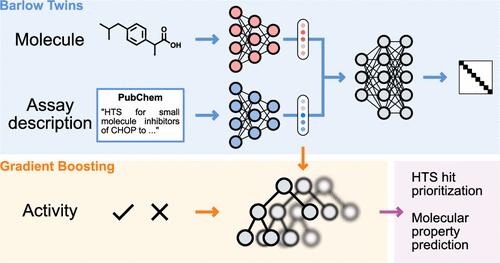
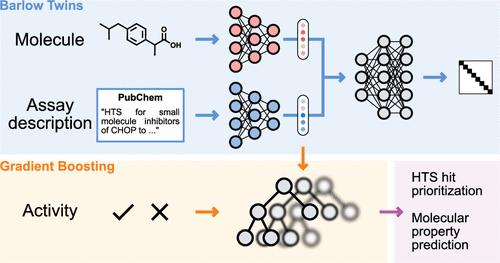
The precise prediction of molecular properties can greatly accelerate the development of new drugs. However, in silico molecular property prediction approaches have been limited so far to assays for which large amounts of data are available. In this study, we develop a new computational approach leveraging both the textual description of the assay of interest and the chemical structure of target compounds. By combining these two sources of information via self-supervised learning, our tool can provide accurate predictions for assays where no measurements are available. Remarkably, our approach achieves state-of-the-art performance on the FS-Mol benchmark for zero-shot prediction, outperforming a wide variety of deep learning approaches. Additionally, we demonstrate how our tool can be used for tailoring screening libraries for the assay of interest, showing promising performance in a retrospective case study on a high-throughput screening campaign. By accelerating the early identification of active molecules in drug discovery and development, this method has the potential to streamline the identification of novel therapeutics.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


