{"title":"Deoxynivalenol induces m<sup>6</sup>A-mediated upregulation of p21 and growth arrest of mouse hippocampal neuron cells in vitro.","authors":"Peirong Xu, Yulan Zhao, Yue Feng, Mindie Zhao, Ruqian Zhao","doi":"10.1007/s10565-024-09872-7","DOIUrl":null,"url":null,"abstract":"<p><p>Hippocampal neurons maintain the ability of proliferation throughout life to support neurogenesis. Deoxynivalenol (DON) is a mycotoxin that exhibits brain toxicity, yet whether and how DON affects hippocampal neurogenesis remains unknown. Here, we use mouse hippocampal neuron cells (HT-22) as a model to illustrate the effects of DON on neuron proliferation and to explore underlying mechanisms. DON exposure significantly inhibits the proliferation of HT-22 cells, which is associated with an up-regulation of cell cycle inhibitor p21 at both mRNA and protein levels. Global and site-specific m<sup>6</sup>A methylation levels on the 3'UTR of p21 mRNA are significantly increased in response to DON treatment, whereas inhibition of m<sup>6</sup>A hypermethylation significantly alleviates DON-induced cell cycle arrest. Further mechanistic studies indicate that the m<sup>6</sup>A readers YTHDF1 and IGF2BP1 are responsible for m<sup>6</sup>A-mediated increase in p21 mRNA stability. Meanwhile, 3'UTR of E3 ubiquitin ligase TRIM21 mRNA is also m<sup>6</sup>A hypermethylated, and another m<sup>6</sup>A reader YTHDF2 binds to the m<sup>6</sup>A sites, leading to decreased TRIM21 mRNA stability. Consequently, TRIM21 suppression impairs ubiquitin-mediated p21 protein degradation. Taken together, m<sup>6</sup>A-mediated upregulation of p21, at both post-transcriptional and post-translational levels, contributes to DON-induced inhibition of hippocampal neuron proliferation. These results may provide new insights for epigenetic therapy of neurodegenerative diseases.</p>","PeriodicalId":9672,"journal":{"name":"Cell Biology and Toxicology","volume":"40 1","pages":"41"},"PeriodicalIF":5.3000,"publicationDate":"2024-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11150311/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Biology and Toxicology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10565-024-09872-7","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
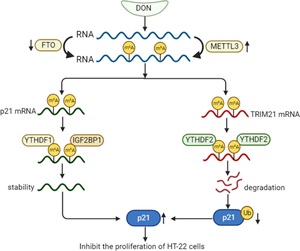
Abstract
Hippocampal neurons maintain the ability of proliferation throughout life to support neurogenesis. Deoxynivalenol (DON) is a mycotoxin that exhibits brain toxicity, yet whether and how DON affects hippocampal neurogenesis remains unknown. Here, we use mouse hippocampal neuron cells (HT-22) as a model to illustrate the effects of DON on neuron proliferation and to explore underlying mechanisms. DON exposure significantly inhibits the proliferation of HT-22 cells, which is associated with an up-regulation of cell cycle inhibitor p21 at both mRNA and protein levels. Global and site-specific m6A methylation levels on the 3'UTR of p21 mRNA are significantly increased in response to DON treatment, whereas inhibition of m6A hypermethylation significantly alleviates DON-induced cell cycle arrest. Further mechanistic studies indicate that the m6A readers YTHDF1 and IGF2BP1 are responsible for m6A-mediated increase in p21 mRNA stability. Meanwhile, 3'UTR of E3 ubiquitin ligase TRIM21 mRNA is also m6A hypermethylated, and another m6A reader YTHDF2 binds to the m6A sites, leading to decreased TRIM21 mRNA stability. Consequently, TRIM21 suppression impairs ubiquitin-mediated p21 protein degradation. Taken together, m6A-mediated upregulation of p21, at both post-transcriptional and post-translational levels, contributes to DON-induced inhibition of hippocampal neuron proliferation. These results may provide new insights for epigenetic therapy of neurodegenerative diseases.
期刊介绍:
Cell Biology and Toxicology (CBT) is an international journal focused on clinical and translational research with an emphasis on molecular and cell biology, genetic and epigenetic heterogeneity, drug discovery and development, and molecular pharmacology and toxicology. CBT has a disease-specific scope prioritizing publications on gene and protein-based regulation, intracellular signaling pathway dysfunction, cell type-specific function, and systems in biomedicine in drug discovery and development. CBT publishes original articles with outstanding, innovative and significant findings, important reviews on recent research advances and issues of high current interest, opinion articles of leading edge science, and rapid communication or reports, on molecular mechanisms and therapies in diseases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


