Hayder A. Al-Atabi, Osamah N. Hasan, Kater alnada faris Husham
{"title":"Advanced Materials Calculations for Methane Dissociation over Ni(111) Surface Using Ab Initio Density Functional Theory","authors":"Hayder A. Al-Atabi, Osamah N. Hasan, Kater alnada faris Husham","doi":"10.1134/s0965544124020014","DOIUrl":null,"url":null,"abstract":"<h3 data-test=\"abstract-sub-heading\">Abstract</h3><p>Advanced materials calculations have been recently widely employed. One of these powerful calculations is Density Functional Theory (DFT). In this work, DFT was used to study the methane dissociation over the surface of the transition metal nickel (Ni) with crystal orientation of (111). The favorable configuration for CH<sub>3</sub> molecule was on the top of Ni with adsorption energy of –2.278 eV, while the face-centered cube position was the favorable structure for the hydrogen (H) atom with a –2.713 eV adsorption energy. The estimated reaction rate constant for the dissociation process was 4.801×10<sup>–15</sup> s<sup>–1</sup>, and the barrier energies were –0.10664191×10<sup>3</sup>, –0.10382003×10<sup>3</sup>, and –0.10616790×10<sup>3</sup> eV for initial, transition, and final state respectively. The adsorption types were physisorption for CH<sub>4</sub> and chemisorption for both CH<sub>3</sub> and H on the Ni(111) surface.</p>","PeriodicalId":725,"journal":{"name":"Petroleum Chemistry","volume":"27 1","pages":""},"PeriodicalIF":1.3000,"publicationDate":"2024-05-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Petroleum Chemistry","FirstCategoryId":"5","ListUrlMain":"https://doi.org/10.1134/s0965544124020014","RegionNum":4,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
引用次数: 0
Abstract
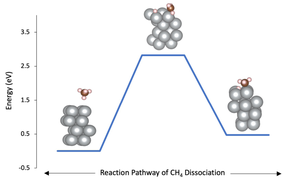
Advanced materials calculations have been recently widely employed. One of these powerful calculations is Density Functional Theory (DFT). In this work, DFT was used to study the methane dissociation over the surface of the transition metal nickel (Ni) with crystal orientation of (111). The favorable configuration for CH3 molecule was on the top of Ni with adsorption energy of –2.278 eV, while the face-centered cube position was the favorable structure for the hydrogen (H) atom with a –2.713 eV adsorption energy. The estimated reaction rate constant for the dissociation process was 4.801×10–15 s–1, and the barrier energies were –0.10664191×103, –0.10382003×103, and –0.10616790×103 eV for initial, transition, and final state respectively. The adsorption types were physisorption for CH4 and chemisorption for both CH3 and H on the Ni(111) surface.
期刊介绍:
Petroleum Chemistry (Neftekhimiya), founded in 1961, offers original papers on and reviews of theoretical and experimental studies concerned with current problems of petroleum chemistry and processing such as chemical composition of crude oils and natural gas liquids; petroleum refining (cracking, hydrocracking, and catalytic reforming); catalysts for petrochemical processes (hydrogenation, isomerization, oxidation, hydroformylation, etc.); activation and catalytic transformation of hydrocarbons and other components of petroleum, natural gas, and other complex organic mixtures; new petrochemicals including lubricants and additives; environmental problems; and information on scientific meetings relevant to these areas.
Petroleum Chemistry publishes articles on these topics from members of the scientific community of the former Soviet Union.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


