{"title":"Design, synthesis and neuroprotective biological evaluation of novel HDAC6 inhibitors incorporating benzothiadiazinyl systems as cap groups","authors":"Bo Han, Xiu Gu, Mengfei Wang, Huihao Wang, Niubing Sun, Xuezhi Yang, Qingwei Zhang","doi":"10.1111/cbdd.14556","DOIUrl":null,"url":null,"abstract":"<p>Histone deacetylase 6 (HDAC6), as the key regulatory enzyme, plays an important role in the development of the nervous system. More and more studies indicate that HDAC6 has become a promising therapeutic target for CNS diseases. Herein we designed and synthesized a series of novel HDAC6 inhibitors with benzothiadiazinyl systems as cap groups and evaluated their activity in vitro and in vivo. Among them, compound <b>3</b> exhibited superior selective inhibitory activity against HDAC6 (IC<sub>50</sub> = 5.1 nM, about 30-fold selectivity over HDAC1). The results of docking showed that compound <b>3</b> can interact well with the key amino acid residues of HDAC6. Compound <b>3</b> showed lower cytotoxicity (20 μM to SH-SY5Y cells, inhibition rate = 25.75%) and better neuroprotective activity against L-glutamate-induced SH-SY5Y cell injury model in vitro. Meanwhile, compound <b>3</b> exhibited weak cardiotoxicity (10 μM hERG inhibition rate = 17.35%) and possess good druggability properties. Especially, compound <b>3</b> could significantly reduce cerebral infarction from 49.87% to 32.18%, and similar with butylphthalide in MCAO model, indicating potential clinical application prospects for alleviating ischemic stroke-induced brain infarction.</p>","PeriodicalId":143,"journal":{"name":"Chemical Biology & Drug Design","volume":null,"pages":null},"PeriodicalIF":3.2000,"publicationDate":"2024-05-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Biology & Drug Design","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cbdd.14556","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
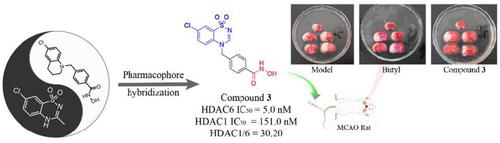
Histone deacetylase 6 (HDAC6), as the key regulatory enzyme, plays an important role in the development of the nervous system. More and more studies indicate that HDAC6 has become a promising therapeutic target for CNS diseases. Herein we designed and synthesized a series of novel HDAC6 inhibitors with benzothiadiazinyl systems as cap groups and evaluated their activity in vitro and in vivo. Among them, compound 3 exhibited superior selective inhibitory activity against HDAC6 (IC50 = 5.1 nM, about 30-fold selectivity over HDAC1). The results of docking showed that compound 3 can interact well with the key amino acid residues of HDAC6. Compound 3 showed lower cytotoxicity (20 μM to SH-SY5Y cells, inhibition rate = 25.75%) and better neuroprotective activity against L-glutamate-induced SH-SY5Y cell injury model in vitro. Meanwhile, compound 3 exhibited weak cardiotoxicity (10 μM hERG inhibition rate = 17.35%) and possess good druggability properties. Especially, compound 3 could significantly reduce cerebral infarction from 49.87% to 32.18%, and similar with butylphthalide in MCAO model, indicating potential clinical application prospects for alleviating ischemic stroke-induced brain infarction.
期刊介绍:
Chemical Biology & Drug Design is a peer-reviewed scientific journal that is dedicated to the advancement of innovative science, technology and medicine with a focus on the multidisciplinary fields of chemical biology and drug design. It is the aim of Chemical Biology & Drug Design to capture significant research and drug discovery that highlights new concepts, insight and new findings within the scope of chemical biology and drug design.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


