A founder mutation in CA5A causing intrafamilial and interfamilial phenotypic variability in a cohort of 18 patients with carbonic anhydrase VA deficiency
IF 1.8 Q2 Biochemistry, Genetics and Molecular Biology
Khalid Al-Thihli, Nadia Al Hashmi, Aaisha Al Balushi, Asila Al-Habsi, Eiman Al-Ajmi, Fatma Al-Jasmi, Fathiya Al-Murshedi
{"title":"A founder mutation in CA5A causing intrafamilial and interfamilial phenotypic variability in a cohort of 18 patients with carbonic anhydrase VA deficiency","authors":"Khalid Al-Thihli, Nadia Al Hashmi, Aaisha Al Balushi, Asila Al-Habsi, Eiman Al-Ajmi, Fatma Al-Jasmi, Fathiya Al-Murshedi","doi":"10.1002/jmd2.12426","DOIUrl":null,"url":null,"abstract":"<p>Carbonic anhydrase VA (CA-VA) deficiency is a rare cause of hyperammonemia caused by biallelic mutations in <i>CA5A.</i> Most patients present with hyperammonemic encephalopathy in early infancy to early childhood, and patients usually have no further recurrence of hyperammonemia with a favorable outcome. This retrospective cohort study reports 18 patients with CA-VA deficiency caused by homozygosity for a founder mutation, c.59G>A p.(Trp20*) in <i>CA5A</i>. The reported patients show significant intrafamilial and interfamilial variability, and display atypical clinical features. Two adult patients were asymptomatic, 7/18 patients had recurrent hyperammonemia, 7/18 patients developed variable degree of developmental delay, 9/11 patients had hyperCKemia, and 7/18 patients had failure to thrive. Microcephaly was seen in three patients and one patient developed a metabolic stroke. The same variant had been reported already in a single South Asian patient presenting with neonatal hyperammonemic encephalopathy and subsequent development of seizures and developmental delay. This report highlights the limitations of current understanding of the pathomechanisms involved in this disorder, and calls for further evaluation of the possible role of genetic modifiers in this condition.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"65 4","pages":"226-232"},"PeriodicalIF":1.8000,"publicationDate":"2024-05-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12426","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12426","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
Abstract
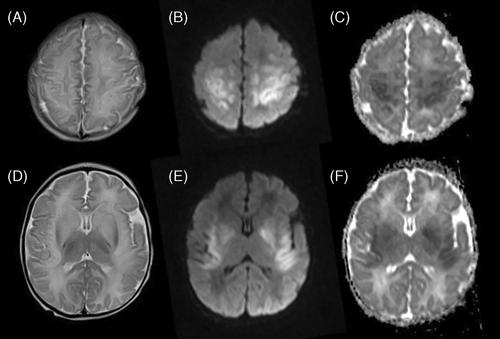
Carbonic anhydrase VA (CA-VA) deficiency is a rare cause of hyperammonemia caused by biallelic mutations in CA5A. Most patients present with hyperammonemic encephalopathy in early infancy to early childhood, and patients usually have no further recurrence of hyperammonemia with a favorable outcome. This retrospective cohort study reports 18 patients with CA-VA deficiency caused by homozygosity for a founder mutation, c.59G>A p.(Trp20*) in CA5A. The reported patients show significant intrafamilial and interfamilial variability, and display atypical clinical features. Two adult patients were asymptomatic, 7/18 patients had recurrent hyperammonemia, 7/18 patients developed variable degree of developmental delay, 9/11 patients had hyperCKemia, and 7/18 patients had failure to thrive. Microcephaly was seen in three patients and one patient developed a metabolic stroke. The same variant had been reported already in a single South Asian patient presenting with neonatal hyperammonemic encephalopathy and subsequent development of seizures and developmental delay. This report highlights the limitations of current understanding of the pathomechanisms involved in this disorder, and calls for further evaluation of the possible role of genetic modifiers in this condition.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


