The neuronal ceroid lipofuscinosis type 2 – associated variants: An analysis of alterations in the TPP1 gene and genotype–phenotype correlation in Ukraine
IF 1.8 Q2 Biochemistry, Genetics and Molecular Biology
{"title":"The neuronal ceroid lipofuscinosis type 2 – associated variants: An analysis of alterations in the TPP1 gene and genotype–phenotype correlation in Ukraine","authors":"Nataliia Olkhovych, Nataliia Pichkur, Nataliia Mytsyk, Rodolfo Tonin, Svitlana Kormoz, Iryna Hregul, Nataliia Samonenko, Tetiana Shklyarskaya, Volodymyr Olkhovych, Olexandr Buryak, Amelia Morrone, Nataliia Gorovenko","doi":"10.1002/jmd2.12423","DOIUrl":null,"url":null,"abstract":"<p>The neuronal ceroid lipofuscinosis type 2 (CLN2) is a heterogeneous group of neurodegenerative lysosomal storage disorders caused by autosomal recessive inheritance of two pathogenic variants in trans in the <i>TPP1</i> gene. Classical late-infantile CLN2 disease has a very well-defined natural history. However, a small number of patients with TPP1 enzyme deficiency present a later onset or protracted disease course within this group there are phenotypic variants. Our work aimed to identify pathological variants in the <i>TPP1</i> gene that conditioned the development of CLN2 disease in Ukrainian patients, to compare these variants with those found in patients from other European and non-European regions, and to make genotype–phenotype associations for this disease. The phenotypes and genotypes of the 48 CLN2-affected individuals belonging to 43 families were profiled through clinical data collection, enzyme analysis, and genotyping. In most patients, genotype and phenotype correlation are in keeping with the data of previous studies. The clinical signs of the disease in patients with new, previously undescribed variants, allowed us to augment existing data about genotype–phenotype correlations for CLN2 disease. The combination of genotype and clinical form of the disease demonstrated that predicting the type and clinical course of the disease based on genotype is very complicated. The data we obtained supplements existing information on genotype–phenotypic correlations in this rare disease, which, in turn, lays the foundation for a personalized approach to the management of this disease.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"65 4","pages":"272-279"},"PeriodicalIF":1.8000,"publicationDate":"2024-05-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12423","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12423","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
Abstract
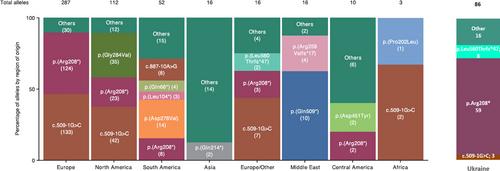
The neuronal ceroid lipofuscinosis type 2 (CLN2) is a heterogeneous group of neurodegenerative lysosomal storage disorders caused by autosomal recessive inheritance of two pathogenic variants in trans in the TPP1 gene. Classical late-infantile CLN2 disease has a very well-defined natural history. However, a small number of patients with TPP1 enzyme deficiency present a later onset or protracted disease course within this group there are phenotypic variants. Our work aimed to identify pathological variants in the TPP1 gene that conditioned the development of CLN2 disease in Ukrainian patients, to compare these variants with those found in patients from other European and non-European regions, and to make genotype–phenotype associations for this disease. The phenotypes and genotypes of the 48 CLN2-affected individuals belonging to 43 families were profiled through clinical data collection, enzyme analysis, and genotyping. In most patients, genotype and phenotype correlation are in keeping with the data of previous studies. The clinical signs of the disease in patients with new, previously undescribed variants, allowed us to augment existing data about genotype–phenotype correlations for CLN2 disease. The combination of genotype and clinical form of the disease demonstrated that predicting the type and clinical course of the disease based on genotype is very complicated. The data we obtained supplements existing information on genotype–phenotypic correlations in this rare disease, which, in turn, lays the foundation for a personalized approach to the management of this disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


