{"title":"Characterization of a novel HDAC2 pathogenetic variant: a missing puzzle piece for chromatinopathies.","authors":"Elisabetta Di Fede, Antonella Lettieri, Esi Taci, Silvia Castiglioni, Stefano Rebellato, Chiara Parodi, Elisa Adele Colombo, Paolo Grazioli, Federica Natacci, Paola Marchisio, Lidia Pezzani, Grazia Fazio, Donatella Milani, Valentina Massa, Cristina Gervasini","doi":"10.1007/s00439-024-02675-0","DOIUrl":null,"url":null,"abstract":"<p><p>Histone deacetylases (HDACs) are enzymes pivotal for histone modification (i.e. acetylation marks removal), chromatin accessibility and gene expression regulation. Class I HDACs (including HDAC1, 2, 3, 8) are ubiquitously expressed and they often participate in multi-molecular protein complexes. To date, three neurodevelopmental disorders caused by mutations in genes encoding for HDACs (HDAC4, HDAC6 and HDAC8) and thus belonging to the group of chromatinopathies, have been described. We performed whole exome sequencing (WES) for a patient (#249) clinically diagnosed with the chromatinopathy Rubinstein-Taybi syndrome (RSTS) but negative for mutations in RSTS genes, identifying a de novo frameshift variant in HDAC2 gene. We then investigated its molecular effects in lymphoblastoid cell lines (LCLs) derived from the patient compared to LCLs from healthy donors (HD). As the variant was predicted to be likely pathogenetic and to affect the sequence of nuclear localization signal, we performed immunocytochemistry and lysates fractionation, observing a nuclear mis-localization of HDAC2 compared to HD LCLs. In addition, HDAC2 total protein abundance resulted altered in patient, and we found that newly identified variant in HDAC2 affects also acetylation levels, with significant difference in acetylation pattern among patient #249, HD and RSTS cells and in expression of a known molecular target. Remarkably, RNA-seq performed on #249, HD and RSTS cells shows differentially expressed genes (DEGs) common to #249 and RSTS. Interestingly, our reported patient was clinically diagnosed with RSTS, a chromatinopathy which known causative genes encode for enzymes antagonizing HDACs. These results support the role of HDAC2 as causative gene for chromatinopathies, strengthening the genotype-phenotype correlations in this relevant group of disorders.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"747-759"},"PeriodicalIF":3.8000,"publicationDate":"2024-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11186948/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-024-02675-0","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/5/16 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
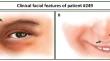
Abstract
Histone deacetylases (HDACs) are enzymes pivotal for histone modification (i.e. acetylation marks removal), chromatin accessibility and gene expression regulation. Class I HDACs (including HDAC1, 2, 3, 8) are ubiquitously expressed and they often participate in multi-molecular protein complexes. To date, three neurodevelopmental disorders caused by mutations in genes encoding for HDACs (HDAC4, HDAC6 and HDAC8) and thus belonging to the group of chromatinopathies, have been described. We performed whole exome sequencing (WES) for a patient (#249) clinically diagnosed with the chromatinopathy Rubinstein-Taybi syndrome (RSTS) but negative for mutations in RSTS genes, identifying a de novo frameshift variant in HDAC2 gene. We then investigated its molecular effects in lymphoblastoid cell lines (LCLs) derived from the patient compared to LCLs from healthy donors (HD). As the variant was predicted to be likely pathogenetic and to affect the sequence of nuclear localization signal, we performed immunocytochemistry and lysates fractionation, observing a nuclear mis-localization of HDAC2 compared to HD LCLs. In addition, HDAC2 total protein abundance resulted altered in patient, and we found that newly identified variant in HDAC2 affects also acetylation levels, with significant difference in acetylation pattern among patient #249, HD and RSTS cells and in expression of a known molecular target. Remarkably, RNA-seq performed on #249, HD and RSTS cells shows differentially expressed genes (DEGs) common to #249 and RSTS. Interestingly, our reported patient was clinically diagnosed with RSTS, a chromatinopathy which known causative genes encode for enzymes antagonizing HDACs. These results support the role of HDAC2 as causative gene for chromatinopathies, strengthening the genotype-phenotype correlations in this relevant group of disorders.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


