Understanding bacterial pathogen diversity: A proteogenomic analysis and use of an array of genome assemblies to identify novel virulence factors of the honey bee bacterial pathogen Paenibacillus larvae
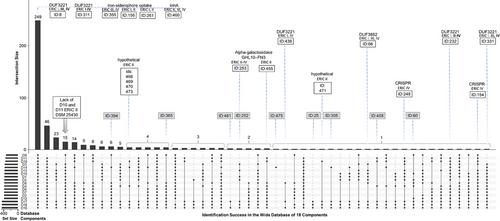
{"title":"Understanding bacterial pathogen diversity: A proteogenomic analysis and use of an array of genome assemblies to identify novel virulence factors of the honey bee bacterial pathogen Paenibacillus larvae","authors":"Tomas Erban, Bruno Sopko","doi":"10.1002/pmic.202300280","DOIUrl":null,"url":null,"abstract":"<p>Mass spectrometry proteomics data are typically evaluated against publicly available annotated sequences, but the proteogenomics approach is a useful alternative. A single genome is commonly utilized in custom proteomic and proteogenomic data analysis. We pose the question of whether utilizing numerous different genome assemblies in a search database would be beneficial. We reanalyzed raw data from the exoprotein fraction of four reference Enterobacterial Repetitive Intergenic Consensus (ERIC) I–IV genotypes of the honey bee bacterial pathogen <i>Paenibacillus larvae</i> and evaluated them against three reference databases (from NCBI-protein, RefSeq, and UniProt) together with an array of protein sequences generated by six-frame direct translation of 15 genome assemblies from GenBank. The wide search yielded 453 protein hits/groups, which UpSet analysis categorized into 50 groups based on the success of protein identification by the 18 database components. Nine hits that were not identified by a unique peptide were not considered for marker selection, which discarded the only protein that was not identified by the reference databases. We propose that the variability in successful identifications between genome assemblies is useful for marker mining. The results suggest that various strains of <i>P. larvae</i> can exhibit specific traits that set them apart from the established genotypes ERIC I–V.</p>","PeriodicalId":224,"journal":{"name":"Proteomics","volume":"24 14","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-05-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/pmic.202300280","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomics","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/pmic.202300280","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Mass spectrometry proteomics data are typically evaluated against publicly available annotated sequences, but the proteogenomics approach is a useful alternative. A single genome is commonly utilized in custom proteomic and proteogenomic data analysis. We pose the question of whether utilizing numerous different genome assemblies in a search database would be beneficial. We reanalyzed raw data from the exoprotein fraction of four reference Enterobacterial Repetitive Intergenic Consensus (ERIC) I–IV genotypes of the honey bee bacterial pathogen Paenibacillus larvae and evaluated them against three reference databases (from NCBI-protein, RefSeq, and UniProt) together with an array of protein sequences generated by six-frame direct translation of 15 genome assemblies from GenBank. The wide search yielded 453 protein hits/groups, which UpSet analysis categorized into 50 groups based on the success of protein identification by the 18 database components. Nine hits that were not identified by a unique peptide were not considered for marker selection, which discarded the only protein that was not identified by the reference databases. We propose that the variability in successful identifications between genome assemblies is useful for marker mining. The results suggest that various strains of P. larvae can exhibit specific traits that set them apart from the established genotypes ERIC I–V.
期刊介绍:
PROTEOMICS is the premier international source for information on all aspects of applications and technologies, including software, in proteomics and other "omics". The journal includes but is not limited to proteomics, genomics, transcriptomics, metabolomics and lipidomics, and systems biology approaches. Papers describing novel applications of proteomics and integration of multi-omics data and approaches are especially welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


