Use of pure recombinant human enzymes to assess the disease-causing potential of missense mutations in urea cycle disorders, applied to N-acetylglutamate synthase deficiency
Nadine Gougeard, Enea Sancho-Vaello, M. Leonor Fernández-Murga, Borja Martínez-Sinisterra, Badr Loukili-Hassani, Johannes Häberle, Clara Marco-Marín, Vicente Rubio
{"title":"Use of pure recombinant human enzymes to assess the disease-causing potential of missense mutations in urea cycle disorders, applied to N-acetylglutamate synthase deficiency","authors":"Nadine Gougeard, Enea Sancho-Vaello, M. Leonor Fernández-Murga, Borja Martínez-Sinisterra, Badr Loukili-Hassani, Johannes Häberle, Clara Marco-Marín, Vicente Rubio","doi":"10.1002/jimd.12747","DOIUrl":null,"url":null,"abstract":"<p><i>N</i>-acetylglutamate synthase (NAGS) makes acetylglutamate, the essential activator of the first, regulatory enzyme of the urea cycle, carbamoyl phosphate synthetase 1 (CPS1). NAGS deficiency (NAGSD) and CPS1 deficiency (CPS1D) present identical phenotypes. However, they must be distinguished, because NAGSD is cured by substitutive therapy with the <i>N</i>-acetyl-L-glutamate analogue <i>N</i>-carbamyl-L-glutamate, while curative therapy of CPS1D requires liver transplantation. Since their differentiation is done genetically, it is important to ascertain the disease-causing potential of <i>CPS1</i> and <i>NAGS</i> genetic variants. With this goal, we previously carried out site-directed mutagenesis studies with pure recombinant human CPS1. We could not do the same with human NAGS (HuNAGS) because of enzyme instability, leading to our prior utilization of a bacterial NAGS as an imperfect surrogate of HuNAGS. We now use genuine HuNAGS, stabilized as a chimera of its conserved domain (cHuNAGS) with the maltose binding protein (MBP), and produced in <i>Escherichia coli</i>. MBP-cHuNAGS linker cleavage allowed assessment of the enzymatic properties and thermal stability of cHuNAGS, either wild-type or hosting each one of 23 nonsynonymous single-base changes found in NAGSD patients. For all but one change, disease causation was accounted by the enzymatic alterations identified, including, depending on the variant, loss of arginine activation, increased K<sub>m</sub><sup>Glutamate</sup>, active site inactivation, decreased thermal stability, and protein misfolding. Our present approach outperforms experimental in vitro use of bacterial NAGS or in silico utilization of prediction servers (including AlphaMissense), illustrating with HuNAGS the value for UCDs of using recombinant enzymes for assessing disease-causation and molecular pathogenesis, and for therapeutic guidance.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 6","pages":"1194-1212"},"PeriodicalIF":4.2000,"publicationDate":"2024-05-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12747","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12747","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
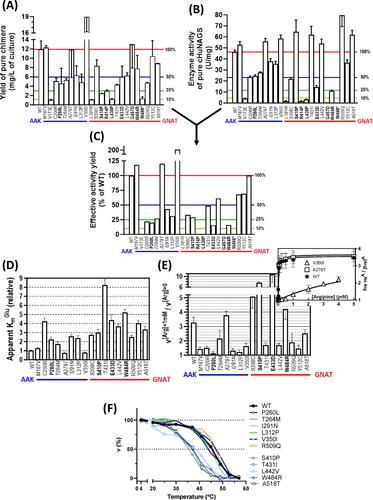
N-acetylglutamate synthase (NAGS) makes acetylglutamate, the essential activator of the first, regulatory enzyme of the urea cycle, carbamoyl phosphate synthetase 1 (CPS1). NAGS deficiency (NAGSD) and CPS1 deficiency (CPS1D) present identical phenotypes. However, they must be distinguished, because NAGSD is cured by substitutive therapy with the N-acetyl-L-glutamate analogue N-carbamyl-L-glutamate, while curative therapy of CPS1D requires liver transplantation. Since their differentiation is done genetically, it is important to ascertain the disease-causing potential of CPS1 and NAGS genetic variants. With this goal, we previously carried out site-directed mutagenesis studies with pure recombinant human CPS1. We could not do the same with human NAGS (HuNAGS) because of enzyme instability, leading to our prior utilization of a bacterial NAGS as an imperfect surrogate of HuNAGS. We now use genuine HuNAGS, stabilized as a chimera of its conserved domain (cHuNAGS) with the maltose binding protein (MBP), and produced in Escherichia coli. MBP-cHuNAGS linker cleavage allowed assessment of the enzymatic properties and thermal stability of cHuNAGS, either wild-type or hosting each one of 23 nonsynonymous single-base changes found in NAGSD patients. For all but one change, disease causation was accounted by the enzymatic alterations identified, including, depending on the variant, loss of arginine activation, increased KmGlutamate, active site inactivation, decreased thermal stability, and protein misfolding. Our present approach outperforms experimental in vitro use of bacterial NAGS or in silico utilization of prediction servers (including AlphaMissense), illustrating with HuNAGS the value for UCDs of using recombinant enzymes for assessing disease-causation and molecular pathogenesis, and for therapeutic guidance.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


