Hosein Fooladi, Steffen Hirte and Johannes Kirchmair*,
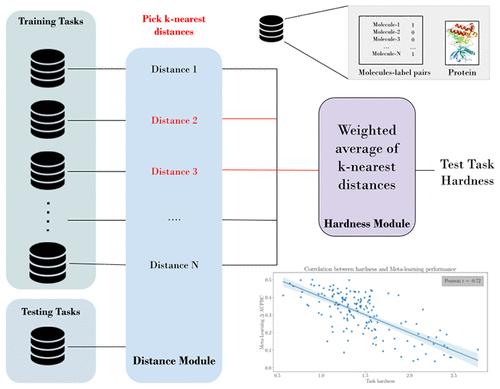
{"title":"Quantifying the Hardness of Bioactivity Prediction Tasks for Transfer Learning","authors":"Hosein Fooladi, Steffen Hirte and Johannes Kirchmair*, ","doi":"10.1021/acs.jcim.4c00160","DOIUrl":null,"url":null,"abstract":"<p >Today, machine learning methods are widely employed in drug discovery. However, the chronic lack of data continues to hamper their further development, validation, and application. Several modern strategies aim to mitigate the challenges associated with data scarcity by learning from data on related tasks. These knowledge-sharing approaches encompass transfer learning, multitask learning, and meta-learning. A key question remaining to be answered for these approaches is about the extent to which their performance can benefit from the relatedness of available source (training) tasks; in other words, how difficult (“hard”) a test task is to a model, given the available source tasks. This study introduces a new method for quantifying and predicting the hardness of a bioactivity prediction task based on its relation to the available training tasks. The approach involves the generation of protein and chemical representations and the calculation of distances between the bioactivity prediction task and the available training tasks. In the example of meta-learning on the FS-Mol data set, we demonstrate that the proposed task hardness metric is inversely correlated with performance (Pearson’s correlation coefficient <i>r</i> = −0.72). The metric will be useful in estimating the task-specific gain in performance that can be achieved through meta-learning.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 10","pages":"4031–4046"},"PeriodicalIF":5.3000,"publicationDate":"2024-05-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jcim.4c00160","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00160","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
Today, machine learning methods are widely employed in drug discovery. However, the chronic lack of data continues to hamper their further development, validation, and application. Several modern strategies aim to mitigate the challenges associated with data scarcity by learning from data on related tasks. These knowledge-sharing approaches encompass transfer learning, multitask learning, and meta-learning. A key question remaining to be answered for these approaches is about the extent to which their performance can benefit from the relatedness of available source (training) tasks; in other words, how difficult (“hard”) a test task is to a model, given the available source tasks. This study introduces a new method for quantifying and predicting the hardness of a bioactivity prediction task based on its relation to the available training tasks. The approach involves the generation of protein and chemical representations and the calculation of distances between the bioactivity prediction task and the available training tasks. In the example of meta-learning on the FS-Mol data set, we demonstrate that the proposed task hardness metric is inversely correlated with performance (Pearson’s correlation coefficient r = −0.72). The metric will be useful in estimating the task-specific gain in performance that can be achieved through meta-learning.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


