Vikas Kumar , Pooja Singh , Shraddha Parate , Rajender Singh , Hyeon-Su Ro , Kyoung Seob Song , Keun Woo Lee , Yeong-Min Park
{"title":"Computational insights into allosteric inhibition of focal adhesion kinase: A combined pharmacophore modeling and molecular dynamics approach","authors":"Vikas Kumar , Pooja Singh , Shraddha Parate , Rajender Singh , Hyeon-Su Ro , Kyoung Seob Song , Keun Woo Lee , Yeong-Min Park","doi":"10.1016/j.jmgm.2024.108789","DOIUrl":null,"url":null,"abstract":"<div><p>Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase that modulates integrin and growth factor signaling pathways and is implicated in cancer cell migration, proliferation, and survival. Over the past decade various, FAK kinase, FERM, and FAT domain inhibitors have been reported and a few kinase domain inhibitors are under clinical consideration. However, few of them were identified as multikinase inhibitors. In kinase drug design selectivity is always a point of concern, to improve selectivity allosteric inhibitor development is the best choice. The current research utilized a pharmacophore modeling (PM) approach to identify novel allosteric inhibitors of FAK. The all-available allosteric inhibitor bound 3D structures with PDB ids 4EBV, 4EBW, and 4I4F were utilized for the pharmacophore modeling. The validated PM models were utilized to map a database of 770,550 compounds prepared from ZINC, EXIMED, SPECS, ASINEX, and InterBioScreen, aiming to identify potential allosteric inhibitors. The obtained compounds from screening step were forwarded to molecular docking (MD) for the prediction of binding orientation inside the allosteric site and the results were evaluated with the known FAK allosteric inhibitor (REF). Finally, 14 FAK-inhibitor complexes were selected from the docking study and were studied under molecular dynamics simulations (MDS) for 500 ns. The complexes were ranked according to binding free energy (BFE) and those demonstrated higher affinity for allosteric site of FAK than REF inhibitors were selected. The selected complexes were further analyzed for intermolecular interactions and finally, three potential allosteric inhibitor candidates for the inhibition of FAK protein were identified. We believe that identified scaffolds may help in drug development against FAK as an anticancer agent.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"130 ","pages":"Article 108789"},"PeriodicalIF":2.7000,"publicationDate":"2024-05-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324000895","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
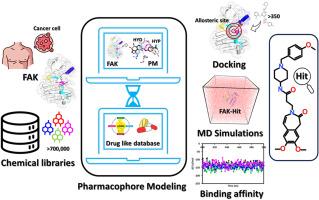
Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase that modulates integrin and growth factor signaling pathways and is implicated in cancer cell migration, proliferation, and survival. Over the past decade various, FAK kinase, FERM, and FAT domain inhibitors have been reported and a few kinase domain inhibitors are under clinical consideration. However, few of them were identified as multikinase inhibitors. In kinase drug design selectivity is always a point of concern, to improve selectivity allosteric inhibitor development is the best choice. The current research utilized a pharmacophore modeling (PM) approach to identify novel allosteric inhibitors of FAK. The all-available allosteric inhibitor bound 3D structures with PDB ids 4EBV, 4EBW, and 4I4F were utilized for the pharmacophore modeling. The validated PM models were utilized to map a database of 770,550 compounds prepared from ZINC, EXIMED, SPECS, ASINEX, and InterBioScreen, aiming to identify potential allosteric inhibitors. The obtained compounds from screening step were forwarded to molecular docking (MD) for the prediction of binding orientation inside the allosteric site and the results were evaluated with the known FAK allosteric inhibitor (REF). Finally, 14 FAK-inhibitor complexes were selected from the docking study and were studied under molecular dynamics simulations (MDS) for 500 ns. The complexes were ranked according to binding free energy (BFE) and those demonstrated higher affinity for allosteric site of FAK than REF inhibitors were selected. The selected complexes were further analyzed for intermolecular interactions and finally, three potential allosteric inhibitor candidates for the inhibition of FAK protein were identified. We believe that identified scaffolds may help in drug development against FAK as an anticancer agent.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


