Immune-related gene methylation prognostic instrument for stratification and targeted treatment of ovarian cancer patients toward advanced 3PM approach
{"title":"Immune-related gene methylation prognostic instrument for stratification and targeted treatment of ovarian cancer patients toward advanced 3PM approach","authors":"Wenshuang Jia, Na Li, Jingjing Wang, Xiaoxia Gong, Serge Yannick Ouedraogo, Yan Wang, Junkai Zhao, Godfrey Grech, Liang Chen, Xianquan Zhan","doi":"10.1007/s13167-024-00359-3","DOIUrl":null,"url":null,"abstract":"<h3 data-test=\"abstract-sub-heading\">Background</h3><p>DNA methylation is an important mechanism in epigenetics, which can change the transcription ability of genes and is closely related to the pathogenesis of ovarian cancer (OC). We hypothesize that DNA methylation is significantly different in OCs compared to controls. Specific DNA methylation status can be used as a biomarker of OC, and targeted drugs targeting these methylation patterns and DNA methyltransferase may have better therapeutic effects. Studying the key DNA methylation sites of immune-related genes (IRGs) in OC patients and studying the effects of these methylation sites on the immune microenvironment may provide a new method for further exploring the pathogenesis of OC, realizing early detection and effective monitoring of OC, identifying effective biomarkers of DNA methylation subtypes and drug targets, improving the efficacy of targeted drugs or overcoming drug resistance, and better applying it to predictive diagnosis, prevention, and personalized medicine (PPPM; 3PM) of OC.</p><h3 data-test=\"abstract-sub-heading\">Method</h3><p>Hypermethylated subtypes (cluster 1) and hypomethylated subtypes (cluster 2) were established in OCs based on the abundance of different methylation sites in IRGs. The differences in immune score, immune checkpoints, immune cells, and overall survival were analyzed between different methylation subtypes in OC samples. The significant pathways, gene ontology (GO), and protein-protein interaction (PPI) network of the identified methylation sites in IRGs were enriched. In addition, the immune-related methylation signature was constructed with multiple regression analysis. A methylation site model based on IRGs was constructed and verified.</p><h3 data-test=\"abstract-sub-heading\">Results</h3><p>A total of 120 IRGs with 142 differentially methylated sites (DMSs) were identified. The DMSs were clustered into a high-level methylation group (cluster 1) and a low-level methylation group (cluster 2). The significant pathways and GO analysis showed many immune-related and cancer-associated enrichments. A methylation site signature based on IRGs was constructed, including RORC|cg25112191, S100A13|cg14467840, TNF|cg04425624, RLN2|cg03679581, and IL1RL2|cg22797169. The methylation sites of all five genes showed hypomethylation in OC, and there were statistically significant differences among RORC|cg25112191, S100A13|cg14467840, and TNF|cg04425624 (<i>p</i> < 0.05). This prognostic model based on low-level methylation and high-level methylation groups was significantly linked to the immune microenvironment as well as overall survival in OC.</p><h3 data-test=\"abstract-sub-heading\">Conclusions</h3><p>This study provided different methylation subtypes for OC patients according to the methylation sites of IRGs. In addition, it helps establish a relationship between methylation and the immune microenvironment, which showed specific differences in biological signaling pathways, genomic changes, and immune mechanisms within the two subgroups. These data provide ones to deeply understand the mechanism of immune-related methylation genes on the occurrence and development of OC. The methylation-site signature is also to establish new possibilities for OC therapy. These data are a precious resource for stratification and targeted treatment of OC patients toward an advanced 3PM approach.</p>","PeriodicalId":54292,"journal":{"name":"Epma Journal","volume":"1 1","pages":""},"PeriodicalIF":5.9000,"publicationDate":"2024-04-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Epma Journal","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s13167-024-00359-3","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
Background
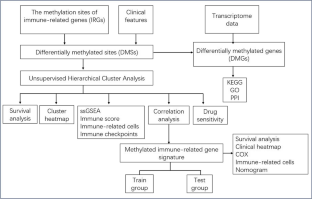
DNA methylation is an important mechanism in epigenetics, which can change the transcription ability of genes and is closely related to the pathogenesis of ovarian cancer (OC). We hypothesize that DNA methylation is significantly different in OCs compared to controls. Specific DNA methylation status can be used as a biomarker of OC, and targeted drugs targeting these methylation patterns and DNA methyltransferase may have better therapeutic effects. Studying the key DNA methylation sites of immune-related genes (IRGs) in OC patients and studying the effects of these methylation sites on the immune microenvironment may provide a new method for further exploring the pathogenesis of OC, realizing early detection and effective monitoring of OC, identifying effective biomarkers of DNA methylation subtypes and drug targets, improving the efficacy of targeted drugs or overcoming drug resistance, and better applying it to predictive diagnosis, prevention, and personalized medicine (PPPM; 3PM) of OC.
Method
Hypermethylated subtypes (cluster 1) and hypomethylated subtypes (cluster 2) were established in OCs based on the abundance of different methylation sites in IRGs. The differences in immune score, immune checkpoints, immune cells, and overall survival were analyzed between different methylation subtypes in OC samples. The significant pathways, gene ontology (GO), and protein-protein interaction (PPI) network of the identified methylation sites in IRGs were enriched. In addition, the immune-related methylation signature was constructed with multiple regression analysis. A methylation site model based on IRGs was constructed and verified.
Results
A total of 120 IRGs with 142 differentially methylated sites (DMSs) were identified. The DMSs were clustered into a high-level methylation group (cluster 1) and a low-level methylation group (cluster 2). The significant pathways and GO analysis showed many immune-related and cancer-associated enrichments. A methylation site signature based on IRGs was constructed, including RORC|cg25112191, S100A13|cg14467840, TNF|cg04425624, RLN2|cg03679581, and IL1RL2|cg22797169. The methylation sites of all five genes showed hypomethylation in OC, and there were statistically significant differences among RORC|cg25112191, S100A13|cg14467840, and TNF|cg04425624 (p < 0.05). This prognostic model based on low-level methylation and high-level methylation groups was significantly linked to the immune microenvironment as well as overall survival in OC.
Conclusions
This study provided different methylation subtypes for OC patients according to the methylation sites of IRGs. In addition, it helps establish a relationship between methylation and the immune microenvironment, which showed specific differences in biological signaling pathways, genomic changes, and immune mechanisms within the two subgroups. These data provide ones to deeply understand the mechanism of immune-related methylation genes on the occurrence and development of OC. The methylation-site signature is also to establish new possibilities for OC therapy. These data are a precious resource for stratification and targeted treatment of OC patients toward an advanced 3PM approach.
期刊介绍:
PMA Journal is a journal of predictive, preventive and personalized medicine (PPPM). The journal provides expert viewpoints and research on medical innovations and advanced healthcare using predictive diagnostics, targeted preventive measures and personalized patient treatments. The journal is indexed by PubMed, Embase and Scopus.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


