{"title":"Alternate conformations found in protein structures implies biological functions: A case study using cyclophilin A","authors":"Chandrasekaran Palaniappan , Santhosh Rajendran , Kanagaraj Sekar","doi":"10.1016/j.crstbi.2024.100145","DOIUrl":null,"url":null,"abstract":"<div><p>Protein dynamics linked to numerous biomolecular functions, such as ligand binding, allosteric regulation, and catalysis, must be better understood at the atomic level. Reactive atoms of key residues drive a repertoire of biomolecular functions by flipping between alternate conformations or conformational substates, seldom found in protein structures. Probing such sparsely sampled alternate conformations would provide mechanistic insight into many biological functions. We are therefore interested in evaluating the instance of amino acids adopted alternate conformations, either in backbone or side-chain atoms or in both. Accordingly, over 70000 protein structures appear to contain alternate conformations only 'A' and 'B' for any atom, particularly the instance of amino acids that adopted alternate conformations are more for Arg, Cys, Met, and Ser than others. The resulting protein structure analysis depicts that amino acids with alternate conformations are mainly found in the helical and β-regions and are often seen in high-resolution X-ray crystal structures. Furthermore, a case study on human cyclophilin A (CypA) was performed to explain the pre-existing intrinsic dynamics of catalytically critical residues from the CypA and how such intrinsic dynamics perturbed upon Ser99Thr mutation using molecular dynamics simulations on the ns-μs timescale. Simulation results demonstrated that the Ser99Thr mutation had impaired the alternate conformations or the catalytically productive micro-environment of Phe113, mimicking the experimentally observed perturbation captured by X-ray crystallography. In brief, a deeper comprehension of alternate conformations adopted by the amino acids may shed light on the interplay between protein structure, dynamics, and function.</p></div>","PeriodicalId":10870,"journal":{"name":"Current Research in Structural Biology","volume":"7 ","pages":"Article 100145"},"PeriodicalIF":2.7000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2665928X24000229/pdfft?md5=99f78944ded3d8b7139bf3f324f58946&pid=1-s2.0-S2665928X24000229-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Research in Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2665928X24000229","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
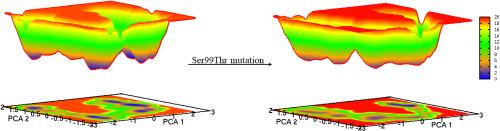
Protein dynamics linked to numerous biomolecular functions, such as ligand binding, allosteric regulation, and catalysis, must be better understood at the atomic level. Reactive atoms of key residues drive a repertoire of biomolecular functions by flipping between alternate conformations or conformational substates, seldom found in protein structures. Probing such sparsely sampled alternate conformations would provide mechanistic insight into many biological functions. We are therefore interested in evaluating the instance of amino acids adopted alternate conformations, either in backbone or side-chain atoms or in both. Accordingly, over 70000 protein structures appear to contain alternate conformations only 'A' and 'B' for any atom, particularly the instance of amino acids that adopted alternate conformations are more for Arg, Cys, Met, and Ser than others. The resulting protein structure analysis depicts that amino acids with alternate conformations are mainly found in the helical and β-regions and are often seen in high-resolution X-ray crystal structures. Furthermore, a case study on human cyclophilin A (CypA) was performed to explain the pre-existing intrinsic dynamics of catalytically critical residues from the CypA and how such intrinsic dynamics perturbed upon Ser99Thr mutation using molecular dynamics simulations on the ns-μs timescale. Simulation results demonstrated that the Ser99Thr mutation had impaired the alternate conformations or the catalytically productive micro-environment of Phe113, mimicking the experimentally observed perturbation captured by X-ray crystallography. In brief, a deeper comprehension of alternate conformations adopted by the amino acids may shed light on the interplay between protein structure, dynamics, and function.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


