{"title":"Prioritizing genomic variants pathogenicity via DNA, RNA, and protein-level features based on extreme gradient boosting","authors":"Maolin Ding, Ken Chen, Yuedong Yang, Huiying Zhao","doi":"10.1007/s00439-024-02667-0","DOIUrl":null,"url":null,"abstract":"<p>Genetic diseases are mostly implicated with genetic variants, including missense, synonymous, non-sense, and copy number variants. These different kinds of variants are indicated to affect phenotypes in various ways from previous studies. It remains essential but challenging to understand the functional consequences of these genetic variants, especially the noncoding ones, due to the lack of corresponding annotations. While many computational methods have been proposed to identify the risk variants. Most of them have only curated DNA-level and protein-level annotations to predict the pathogenicity of the variants, and others have been restricted to missense variants exclusively. In this study, we have curated DNA-, RNA-, and protein-level features to discriminate disease-causing variants in both coding and noncoding regions, where the features of protein sequences and protein structures have been shown essential for analyzing missense variants in coding regions while the features related to RNA-splicing and RBP binding are significant for variants in noncoding regions and synonymous variants in coding regions. Through the integration of these features, we have formulated the Multi-level feature Genomic Variants Predictor (ML-GVP) using the gradient boosting tree. The method has been trained on more than 400,000 variants in the Sherloc-training set from the 6th critical assessment of genome interpretation with superior performance. The method is one of the two best-performing predictors on the blind test in the Sherloc assessment, and is further confirmed by another independent test dataset of de novo variants.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":"20 1","pages":""},"PeriodicalIF":3.8000,"publicationDate":"2024-04-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-024-02667-0","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
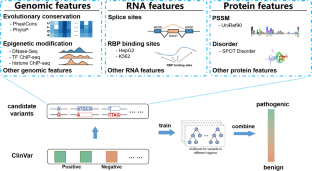
Genetic diseases are mostly implicated with genetic variants, including missense, synonymous, non-sense, and copy number variants. These different kinds of variants are indicated to affect phenotypes in various ways from previous studies. It remains essential but challenging to understand the functional consequences of these genetic variants, especially the noncoding ones, due to the lack of corresponding annotations. While many computational methods have been proposed to identify the risk variants. Most of them have only curated DNA-level and protein-level annotations to predict the pathogenicity of the variants, and others have been restricted to missense variants exclusively. In this study, we have curated DNA-, RNA-, and protein-level features to discriminate disease-causing variants in both coding and noncoding regions, where the features of protein sequences and protein structures have been shown essential for analyzing missense variants in coding regions while the features related to RNA-splicing and RBP binding are significant for variants in noncoding regions and synonymous variants in coding regions. Through the integration of these features, we have formulated the Multi-level feature Genomic Variants Predictor (ML-GVP) using the gradient boosting tree. The method has been trained on more than 400,000 variants in the Sherloc-training set from the 6th critical assessment of genome interpretation with superior performance. The method is one of the two best-performing predictors on the blind test in the Sherloc assessment, and is further confirmed by another independent test dataset of de novo variants.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


