Autism spectrum disorder profiles in RASopathies: A systematic review
IF 1.5
4区 医学
Q4 GENETICS & HEREDITY
引用次数: 0
Abstract
BackgroundRASopathies are associated with an increased risk of autism spectrum disorder (ASD). For neurofibromatosis type 1 (NF1) there is ample evidence for this increased risk, while for other RASopathies this association has been studied less. No specific ASD profile has been delineated so far for RASopathies or a specific RASopathy individually.MethodsWe conducted a systematic review to investigate whether a specific RASopathy is associated with a specific ASD profile, or if RASopathies altogether have a distinct ASD profile compared to idiopathic ASD (iASD). We searched PubMed, Web of Science, and Open Grey for data about ASD features in RASopathies and potential modifiers.ResultsWe included 41 articles on ASD features in NF1, Noonan syndrome (NS), Costello syndrome (CS), and cardio‐facio‐cutaneous syndrome (CFC). Individuals with NF1, NS, CS, and CFC on average have higher ASD symptomatology than healthy controls and unaffected siblings, though less than people with iASD. There is insufficient evidence for a distinct ASD phenotype in RASopathies compared to iASD or when RASopathies are compared with each other. We identified several potentially modifying factors of ASD symptoms in RASopathies.ConclusionsOur systematic review found no convincing evidence for a specific ASD profile in RASopathies compared to iASD, or in a specific RASopathy compared to other RASopathies. However, we identified important limitations in the research literature which may also account for this result. These limitations are discussed and recommendations for future research are formulated.
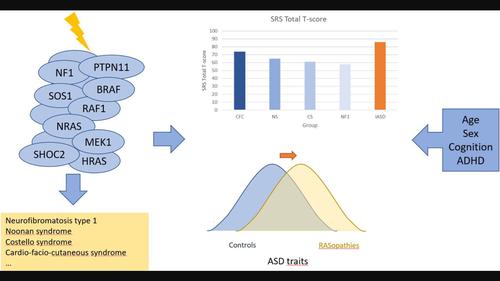
RAS病的自闭症谱系障碍特征:系统综述
背景RAS病与自闭症谱系障碍(ASD)风险增加有关。对于 1 型神经纤维瘤病 (NF1),有大量证据表明这种风险会增加,而对于其他 RAS 病,这种关联的研究较少。我们进行了一项系统性综述,以研究特定的 RAS 病是否与特定的 ASD 特征相关,或者与特发性 ASD(iASD)相比,RAS 病是否具有独特的 ASD 特征。我们在PubMed、Web of Science和Open Grey上搜索了有关RAS病的ASD特征和潜在修饰因素的数据。结果我们收录了41篇有关NF1、努南综合征(NS)、科斯特罗综合征(CS)和心-面-皮肤综合征(CFC)的ASD特征的文章。NF1、NS、CS和CFC患者的ASD症状平均高于健康对照组和未受影响的兄弟姐妹,但低于iASD患者。目前还没有足够的证据表明,与iASD相比,或当RAS病相互比较时,RAS病具有独特的ASD表型。结论我们的系统综述没有发现令人信服的证据表明,与 iASD 相比,或与其他 RAS 病相比,RAS 病具有特定的 ASD 特征。不过,我们也发现了研究文献中的一些重要局限性,这些局限性也可能是造成这一结果的原因。我们对这些局限性进行了讨论,并为今后的研究提出了建议。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Molecular Genetics & Genomic Medicine
Biochemistry, Genetics and Molecular Biology-Genetics
CiteScore
4.20
自引率
0.00%
发文量
241
审稿时长
14 weeks
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


